
Mitochondrial electron transport chain, ROS generation and uncoupling (Review)
- Authors:
- Published online on: May 8, 2019 https://doi.org/10.3892/ijmm.2019.4188
- Pages: 3-15
-
Copyright: © Zhao et al. This is an open access article distributed under the terms of Creative Commons Attribution License [CC BY_NC 4.0].
Abstract
1. Introduction
The chemiosmotic theory proposed by Peter Mitchell (1) in 1961 states that the transfer of electrons derived from substrate oxidation and ATP synthesis are coupled in the mitochondrial ETC, but that does not mean that the transfer of electrons is 100% efficient. Due to the existence of electron leak and proton leak, not all electrons in the ETC can be transferred to the final electron acceptor O2 and the energy released by the transferred electrons cannot be completely coupled with ATP generation. However, both the ROS generated by electron leak and the UCPs implicated in proton leak play an important role in the physiology and pathology of cells. Therefore, it is extremely important to understand the process of electron transfer in the ETC and the mechanism of electron leak and proton leak.
In this review, the basic components of the ETC are discussed and the process of electron transfer in each complex, including the structure, composition and function of each complex is reviewed. In addition, the ROS generation sites in the ETC are summarized and the ROS regulation is mentioned. Moreover, proton leak is emphatically introduced, including the structure, tissue distribution, functions and regulatory factors of UCPs. The diseases implicated in ROS or UCPs are simply summarized.
2. Mitochondrial ETC and ATP synthase
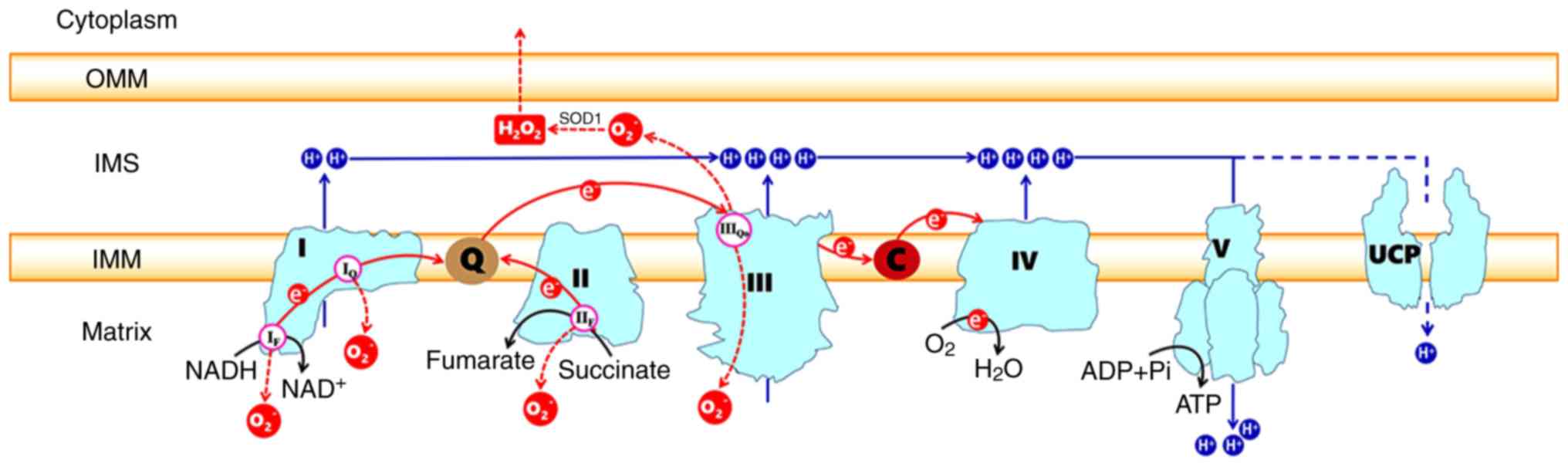
The ETC, which is composed of transmembrane protein complexes (I-IV) and the freely mobile electron transfer carriers ubiquinone and cytochrome c, exists in the folded inner membranes called cristae (Fig. 1). The complexes must be assembled into a specifically configured supercomplex to function properly (2,3). These assembled components together with F1F0ATP synthase (namely, complex V) become the basis of ATP production during oxidative phosphorylation (OXPHOS). To better understand the whole process of how electron transportation produces ATP via the ETC, it is necessary to know the ultrastructure and function of the individual complexes.
Complex I (CI)
CI, also called NADH-ubiquinone oxidoreductase, is the largest multisubunit enzyme complex in the ETC. The key role of CI is to transfer electrons from matrix NADH to ubiquinone, as the name implies. A number of studies have reported the structure of the bacterial mitochondrial CI using X-ray crystallography at a nearly atomic resolution (4,5). Mitochondria from the Bos taurus heart have been regarded as the best model for human CI (6-9). These studies demonstrate that the L-shaped eukaryotic CI contains two domains: The membrane arm embedded in the inner membranes and the matrix arm protruding into the matrix. The two domains are mainly composed of 14 core subunits that are conserved from bacterial CI and are the core of the enzymatic reaction. There are 45 clearly identified proteins that participate in the formation of the core subunits. The matrix arm contains seven core subunits (NDUFS1, NDUFS2, NDUFS3, NDUFS7, NDUFS8, NDUFV1 and NDUFV 2) that contain the following cofac-tors: A flavin mononucleotide (FMN) molecule; 7-9 FeS clusters [including the (2Fe-2S)N1b, (4Fe-4S)N3, (4Fe-4S)N4, (4Fe-4S)N5, (4Fe-4S)N6a/b and (4Fe-4S)N2 clusters] (4,10); and the final electron accepting iron-sulfur cluster (N2 cluster), which was recently found to deliver electrons to ubiquinone binding sites (11). The membrane arm contains seven hydrophobic subunits (ND1-6 and ND4L), all of which are encoded by the mitochondrial genome. In addition, a large number of accessory subunits are arranged around the core subunits. The assembly of these modules has been reviewed in detail elsewhere (12). An FMN bound at the cusp of the matrix arm could form FMNH2 by accepting a pair of electrons derived from matrix NADH, which is primarily produced by the tricarbox-ylic acid (Krebs) cycle that continuously occurs in the matrix. These interactions also mean that electrons go into the ETC and are then passed to ubiquinone via a chain of iron-sulfur clusters arranged from low to high potential [the transfer order was reported as FMN→N3→N1b→N4→N5→N6a→N6b→ N2 (4)]. The ubiquinone binding site is located at the junction of the membrane arm and matrix arm, in which ubiquinone (CoQ) is reduced to ubiquinol (QH2). Then, the conformational changes of the N2 cluster induce the formation of a proton translocation channel by the ND1, ND3, ND6 and ND4L subunits near the CoQ binding site (13). The energy released by the transfer of a pair of electrons from NADH to CoQ in CI probably (not definitively) induce the pumping of four protons from the matrix into the intermembrane space (14-17). Several hypotheses exist in current research: Ohnishi (18) proposed a hypothesis that two protons are indirectly pumped out in a conformation-coupled manner and that the other two protons are directly pumped out by the induction of ubiquinone redox. Sazanov and Hinchliffe (4) hypothesized that three protons are indirectly pumped via three antiporter homologs, and the final proton is shifted in an unclear way. In addition, Tan et al (14) speculated that the conformation changes and the density of water molecules in the trans-membrane domain determine the proton translocation in CI. However, how the energy transfers from the redox reaction to proton translocation are still unknown.
Complex II (CII)
CII, namely, succinate dehydrogenase, is a component of the Krebs cycle as well as the ETC, serving as a link between metabolism and OXPHOS (19,20). As a part of the Krebs cycle, CII catalyzes the oxidation of succinate to fumarate. CII is another entry point for electrons and donates them from succinate to CoQ via FeS clusters, similar to CI. CII consists of four subunits (20). A total of two of the subunits, the membrane-anchor proteins CybL and CybS, are hydrophobic, anchor the complex to the inner membrane, and contain the CoQ binding site. The other two subunits are located on the matrix side of the inner membrane and contain the binding site of the substrate succinate, three FeS clusters [(2Fe-2S), (4Fe-4S) and (3Fe-4S)], and a flavoprotein covalently bound to a FAD cofactor. The assembly steps of the four subunits are detailed elsewhere (21). FAD is reduced to FADH2 after receiving electrons from succinate and then transfers the electrons to FeS clusters. Then, CoQ is reduced to QH2 after obtaining the electrons from the FeS cluster (3Fe-4S) (22). Electron transport in CII is not accompanied by the translocation of protons.
Complex III (CIII)
CIII is commonly referred to as a cytochrome bcl complex, or CoQ-cytochrome c reductase and transfers the electrons carried by QH2 to cytochrome c. CIII is a symmetrical dimer with 11 subunits per monomer (23). The catalytically active subunits are cytochrome b (bL and bH), cytochrome c1 and a high-potential (2Fe-2S) cluster wrapped by an iron-sulphur protein (24). There are two CoQ binding sites on both ends of cytochrome b embedded in the inner membrane of the mitochondria, one of which is the QH2 oxidation site (Qo) located at the cytoplasmic side, which is related to the low potential cytochrome bL. The other is the Q- reduction site (Qi) on the side of the matrix, which is related to the high potential cytochrome bH (25). The electron transfer process of CIII is accomplished by the Q-cycle (24-27). QH2 is oxidized to ubisemiquinone (QH-) after transferring an electron to the (2Fe-2S) cluster and two protons are concurrently released into the mitochondrial intermembrane space (IMS) from the matrix (28). The (2Fe-2S) cluster transfers this electron to cytochrome c1, from which it is transferred to cytochrome c, a mobile electron carrier. Then, the highly reductive QH− formed at the Qo site rapidly transfers the second electron to cytochrome bL, which in turn transfers it to cytochrome bH at the Qi site. Reduced cytochrome bH transfers this electron to the CoQ of the Qi site, forming a QH−. To complete the Q-cycle, the second QH2 molecule is oxidized at the Qo site while displacing the other two protons. Similarly, one electron is transferred to the (2Fe-2S) cluster and the other electron to cytochrome bH and finally to QH− of the Qi site to produce QH2.
Complex IV (CIV)
CIV, also known as cytochrome c oxidase, transfers electrons from cytochrome c to the terminal electron acceptor O2 to generate H2O. Mammalian CIV consists of 13 different subunits containing four redox-active metal centers, namely, CuA, heme a (Fea) and a binuclear center composed of heme a3 (Fea3) and CuB (29,30). Subunits I, II, III are encoded by mitochondrial DNA and are the core subunits, while the 10 nuclear-coded subunits are the accessory subunits (31,32). Subunit I contains three of the four cofactors, heme a and the binuclear center, which transfers electrons from heme a to O2 (29). Subunits II and III are located on both sides of subunit I and there are two CuA cofactors on the side of the intermembrane space of subunit II. Subunit III stabilizes the other two core proteins and is mainly involved in proton pumping (33,34). The nuclear-coded subunits participate in the modulation of physiological activity via the allosteric ATP-mediated inhibition of CIV, which depends on the ATP/ADP-ratio (35-39).
Cytochrome c, similar to CoQ, is a mobile electron carrier that is loosely connected to the outer surface of the inner mitochondrial membrane by electrostatic interactions, allowing it to interact with the cytochrome c1 of CIII and to accept electrons (39). The reduced cytochrome c moves along the surface of the membrane and interacts with subunit II of CIV by electrostatic interactions, simultaneously transmitting electrons to the CuA site of subunit II, and then the electrons are passed from heme a to the binuclear center of subunit I (29,39), where the O2 is reduced to H2O. A total of four electrons at a time from cytochrome c are almost simultaneously transferred to bind dioxygen; eight protons in total are removed from the matrix, of which half are used to form the two water molecules and the other four are pumped across the membrane into the IMS (40).
Complex V (CV)
CV is normally called F1F0 ATP synthase and consists of two functional domains: F0 and F1. The F0 domain, located in the inner mitochondrial membrane, contains a subunit c-ring, including one of each of the subunits a, b, d, F6 and oligomycin sensitivity-conferring protein (OSCP) as well as the accessory subunits e, f, g and A6L (41,42). The subunits b, d, F6 and OSCP form the peripheral stalk, which is located on one side of the complex. A number of additional subunits (e, f, g and A6L), which all span the membrane, are associated with the c-ring subunit. The F1 domain, situated in the mitochondrial matrix, consists of soluble subunits: Three α subunits, three β subunits and one of each of the γ, δ and ε subunits (42,43). The three α and three β subunits make up the catalytic head of F1, and the γ, δ and ε subunits constitute the central stalk that connects the F1 head and F0 subunit c-ring (41,43,44). The ETC transfers two electrons at a time to monooxygen to generate one H2O molecule, which is accompanied by the pumping of four, four and two protons from the matrix to the IMS through CI, CIII, and CIV, respectively (or zero, four and two protons through the CII, CIII, and CIV, respectively). Then, the protons pass from the IMS to the matrix through F0, which transfers the stored energy created by the proton electrochemical gradient to F1, causing a conformational change in F1F0 ATP synthase so that ADP can be phosphorylated to form ATP (41).
In conclusion, the entire composition of each individual complex has been well described over the past century and it is now widely accepted that these complexes must establish interactions and form supercomplexes to perform their function. Due to the application of cryo-electron microscopy, a greater understanding of the high-resolution structure of these complexes has been gained (45-47).
3. ROS generation in the ETC
The sites of ROS production in the ETC
Mitochondria are a main source of cellular ROS. Under physiological conditions, 0.2-2% of the electrons in the ETC do not follow the normal transfer order but instead directly leak out of the ETC and interact with oxygen to produce superoxide or hydrogen peroxide (48,49). A total of 11 sites that produce superoxide (O2−) and/or hydrogen peroxide (H2O2) that are associated with substrate oxidation and the ETC have currently been identified in mammalian mitochondria (50). Sites OF, PF, BF and AF are in the 2-oxoacid dehydrogenase complexes, sites IF and IQ are in CI, site IIIQo is in CIII, and sites IIF, GQ, EF and DQ are linked to the Q-dependent dehydrogenases in the QH2/Q pool (50). The occurrence of numerous diseases and hypoxia are closely related to the increase of ROS production. CI and CIII, especially CI, are considered to be the main sites of ROS production in mitochondria (51,52).
ROS can be generated in the matrix at both site IF (FMN site) and site IQ (CoQ binding site) during the transfer of electrons from NADH to CoQ in CI (Fig. 1). Rotenone and piericidin are site IQ inhibitors that interrupt the electron transfer to CoQ and increase ROS production at site IF. Hernansanz-Agustin et al (53) found that acute hypoxia produces a superoxide burst during the first few minutes in arterial endothelial cells and CI mainly participated in this process.
CII produces ROS at site IIF (Fig. 1), which is associated with succinate dehydrogenase. The level of ROS produced by site IIF under normal conditions is negligible, but the increases in ROS observed in CII mutation-related diseases are mainly derived from site IIF (54,55). The study of isolated mitochondria from rat skeletal muscle also indicated that the maximum capacity for ROS production of site IIF is very high, exceeded only by site IIIQo and perhaps site IQ (50,56). The capacity of site IIF to produce ROS is closely related to the quantity of reduced flavoprotein, whose FAD is a potent site of electron leakage to generate ROS. ROS are exclusively produced in the matrix, because the flavoprotein is located on the matrix side of the inner mitochondrial membrane (56). In addition, any contribution by site IIF can be dampened by the occupation of the CII flavoprotein site by dicarboxylic acids, particularly oxaloacetate, malate and succinate, which blocks the access of oxygen to site IIF, where it would form ROS (21,57).
CIII produces small amounts of ROS, which could be overlooked compared to the ROS production of CI (52). CIII transfers electrons through the Q-cycle. In this process, ubisemiquinone (QH−) of the Qo site carrying a single electron can move freely in CIII, directly leaking the single electron to O2, forming ROS through a nonenzymatic reaction (58,59). The formed ROS can be released into both the matrix and IMS despite the location of the Qo site on the IMS side of the inner mitochondrial membrane. Muller et al (60) built two models explaining how superoxide can reach the matrix. The O2− released into the IMS can be converted to the relatively more stable form of H2O2 by superoxide dismutase (SOD) enzymes (Fig. 1). This permanent and stable oxidant molecule, which freely disperses through the outer membrane of mitochondria, acts as an intracellular signaling molecule, physiologically functioning via the direct modification of amino acids (61). However, supporting evidence demonstrates that O2−can permeate through the mitochondrial membrane into the cytosol through anion channels (62). Treberg et al (63) experiments in the mitochondria of wild-type rat skeletal muscle proved that ~63% of ROS are produced in the matrix. Antimycin A can specifically block the Qi site of CIII, resulting in the stalling of electrons on the QH- at site IIIQo, which could react with O2 to generate ROS (64,65). As specific inhibitors of the Qo site, stigmatellin and myxothiazol can block the binding of QH2 to the Qo site, which also blocks the transfer of electrons into CIII, thereby preventing the production of ROS in CIII (64). Previously, a chemical suppressor of site IIIQo electron leak called S3QELs was screened and found to specifically suppress the ROS formation at site IIIQo without affecting electron transport or the redox states of other centers (66).
CIV is less prone to produce ROS when O2 is bound to Fea32+ or when O2 is negatively polarized (O2−) and expected to undergo a structural change. This structural change allows O2− to receive three electron equivalents from CuB1 +, Fea33+ and the hydroxyl group of Tyr244 (Tyr-OH) in no particular order, providing the complete reduction of O2 and minimizing the production of ROS (67). It is important to note that the binu-clear center structure of CIV is crucial for the nonsequential transfer of the three electron equivalents (39,67).
ROS as signaling molecules in physiological or patho- logical conditions
In the past, it was believed that ROS were exclusively harmful to cells. However, recent studies have demonstrated that ROS appear to be very important second messengers that mediate different intracellular pathways (50,61,68). ROS act through the oxidative modification of numerous types of proteins, particularly receptors, kinases, phosphatases, caspases, ion channels and transcription factors (68). The ROS produced from CIII are necessary for HIF-1α stabilization and consequently, for the proliferation of cells, including vascular smooth muscle cells, endothelial cells and erythroid progenitors (69). There is ample evidence that ROS are also involved in different protein kinase signaling cascades, such as the protein kinase B (AKT), AMP-activate protein kinase (AMPK) and mitogen-activated protein kinase kinase kinase/mitogen-activated protein kinase 8 pathways, changing the fate of cells between autophagy and apoptosis [(Table I and (70)]. Under hypoxic conditions, ROS activate AMPK, which can upregulate cytoprotective autophagy by inhibiting downstream mammalian target of rapamycin activity (71). ROS have also been demonstrated to regulate synaptic plasticity-related signalling molecules, receptors and channels, including N-methyl-d-aspartate receptor (72), Ca2+ channel (73,74), Ca2+ kinase II (CaMKII) (75), extracellular signal-regulated kinase (76) and cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) (74,77). ROS are also necessary for long-term potentiation, a phenomenon of synaptic plasticity widely regarded as one of the main molecular mechanisms that form the basis of learning and memory (77,78). Physiological levels of ROS can promote the establishment of neuronal polarity and regulate neuronal cytoskeletal organization and dynamics by regulating intracellular Ca2+ release (79-81).
Table IThe signaling pathways involved in the different cell fates in which the mitochondrial production of ROS has been implicated. |
The amount of ROS generated as a result of a stimulus determines whether ROS play beneficial or harmful roles, which means different physiological or pathological pathways are activated. A large amount of ROS cause lipid peroxidation, DNA damage, protein oxidation, irreversible impairment of mitochondria, insufficient ATP generation and, eventually, cell death (82). The ROS-mediated activation of NHE-1 is implicated in cardiac hypertrophy (83). In addition, the activation of CaMKII by ROS contributes to an increase in cardiomyocyte death and the development of heart failure (84). ROS are involved in a number of chronic inflammatory diseases, particularly atherogenesis, through activating the NF-κB pathway (85). In addition, it is widely known that the ROS burst during reperfusion plays a critical role in ischemia-reperfusion injury (86). Table II summarizes the pathologies in which ROS has been implicated.
Table IIThe pathologies in which the mitochondrial production of reactive oxygen species has been implicated. |
4. Mitochondrial proton leak
The overview of proton leak
OXPHOS is not completely coupled. Under routine circumstances, a small number of protons do not pass through ATP synthase and instead flow directly into the mitochondrial matrix across the inner mitochondrial membrane, without the generation of ATP, in a process known as proton leak. In the concept of 'respiratory state' proposed by Chance and Williams (16), mitochondrial respiration persists in the absence of ADP (state 4) and reflects the oxygen consumption of proton leak. The existence of proton leak can also be proven by the collapse of the proton gradient (Δp) in the presence of the ATP synthase inhibitor oligomycin in isolated mitochondria (137).
It was found that the proton leak of the inner mitochondrial membrane demonstrated nonohmic conductivity (137). According to Ohm's Law (R=U/I), the resistance of a conductor is fixed and the electric current increases linearly with increasing voltage. However, the rate of proton leak increases exponentially with increasing ΔΨ. That is, the proton conductivity increases when ΔΨ is high. The existence of nonohmic conductivity indicates that there is a bidirectional self-regulatory mechanism between electron transport and proton re-entry: Protons are pumped out of the matrix into the IMS driven by the electron transport in CI, CIII, CIV, and ΔΨ is gradually elevated. On one hand, the elevated ΔΨ inhibits the transfer of electrons to further elevate ΔΨ, through which the inner membrane is protected from electric shock and maintains suitable ΔΨ. On the other hand, the exorbitant ΔΨ can cause the increase in proton leak to decrease.
Proton leak consists of two parts: Basal proton leak and inducible proton leak. Basal proton leakage is not regulated and is related to the lipid bilayer of the inner mitochondrial membrane and the adenine nucleotide translocase (ANT). Induced proton leak is precisely regulated and can be catalyzed or suppressed by uncoupling proteins (UCPs) and ANT.
Basal proton leak has an important relationship with the basal metabolic rate (BMR) in mammals at rest. The lower the BMR of a species, the weaker the basal proton conductance. Studies have demonstrated that the extent of basal proton leak among species has a phylogenetic relationship (138,139). Although the lipid bilayer can significantly increase proton conductivity, only ~5% of basal proton leak is mediated by lipid bilayers (140) and most of the basic proton leak is correlated with ANT (141). UCP1, which is abundant in brown adipose tissue (BAT), may also be involved in basal proton leak (142), although there remains controversy (143). In particular, proton leak through ANT or UCP1 is independent of protein activity, as proton leak still occurs in the presence of the ANT inhibitor carboxyatractylate and the UCP1 inhibitor GDP (141,144).
The majority of the induced proton leak is catalyzed by UCPs. UCPs belong to the family of mitochondrial anion carrier proteins, through which the protons can reflux into the matrix. To date, five UCPs have been identified in mammals, UCP1, UCP2, UCP3, UCP4 and UCP5, and all are present in the form of dimers in the inner mitochondrial membrane (145). These UCPs have a purine nucleotide binding site located on a projection in the IMS (146). The purine nucleotides (ATP, ADP, GTP and GDP) are inhibitors of UCP-mediated proton flux, whereas ROS and fatty acids are activators (147,148). In addition to the role of uncoupling, UCPs may also participate in other processes, such as the regulation of calcium homeostasis, ion transportation or synaptic plasticity (149,150).
UCP1-5
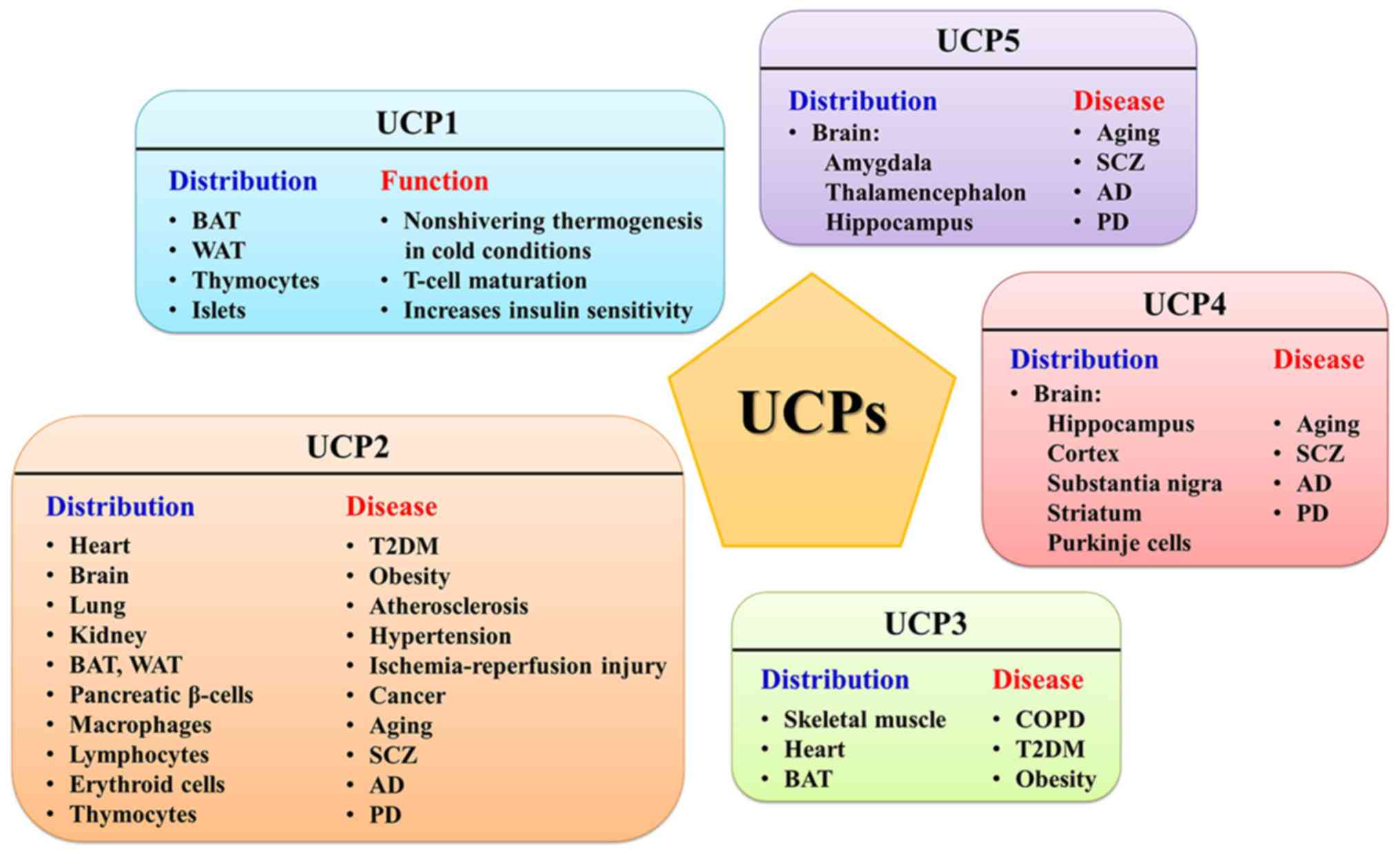
UCP1 is mainly expressed in BAT, which converts stored energy in Δp into heat for thermogenesis (151). UCP1 can also be detected in the beige adipocytes of white adipose tissue (WAT) during thermal acclimation under specific conditions (152). The genetic deletion of UCP1 severely inhibits cold adaptive thermogenesis and diet-induced adrenergic thermogenesis, and UCP1-null mice develop fatal hypothermia upon cold exposure (153,154). Interestingly, WAT can exert nonshivering thermogenesis with a UCP1-independent pathway (155,156). UCP1 has also been found in thymocytes and demonstrated to be involved in the maturation and fate determination of developing T-cells (157-159). Sale et al (160) found that UCP1 is expressed in islets and associates with the acute insulin response to glucose.
UCP1-catalyzed proton leak could be activated by long chain free fatty acids and inhibited by purine nucleotides (161). Acute cold or overfeeding stimuli induce the release of norepinephrine by sympathetic nerves and then induce cAMP-responsive pathways through β3-adrenergic receptors on brown adipocytes, which could further promote the transcription of UCP1 and lipolysis for more free fatty acids (162). There are currently three models for the regulated mechanism of UCP1-implicated proton leak (162-169).
UCP2 and UCP3, paralogues of UCP1, exhibit ~60% sequence identity with UCP1 and ~70% identity with each other (170,171). UCP2 is rather ubiquitous, expressed in WAT, BAT, macrophages, erythroid cells, thymocytes and pancreatic β-cells as well as heart, brain, lung, kidney and lymphocytes (172-176). The UCP3 gene is mainly expressed in skeletal muscle, BAT and heart (177,178) and has also been detected in the thymus, spleen (179) and skin cells (180). Studies with UCP2/3-ablated mice have demonstrated that UCP2 and UCP3 are not implicated in adaptive thermogenesis or the regulation of body weight (170,181). However, the role of UCP2 and UCP3 in inhibiting the production of ROS in mitochondria by reducing ΔΨ is widely accepted (182). A strong correlation between ROS production and mitochondrial membrane potential (ΔΨ) has already been confirmed (183). Experiments have demonstrated that ROS production is increased in UCP2/3-ablated mice (184-186). ROS-induced lipid hydroperoxides such as hydroxynonenal can activate UCP2/3-mediated proton leak, but the mechanism remains uncertain (178). By reducing ΔΨ, the transfer of electrons in the ETC can be accelerated and the likelihood of electrons being directly transferred to O2 can be minimized. Therefore, mild uncoupling is a feedback mechanism adopted by the body to prevent excessive ROS in the mitochondria, which was termed 'uncoupling to survive' (187). However, whether UCP1 is implicated in the regulation of ROS in BAT is still controversial (175,188,189). In addition to the function of reducing the generation of ROS, UCP3 has been demonstrated to be involved in exporting mitochondrial fatty acid anions to the cytoplasm, thereby protecting the mitochondrial against lipid peroxide-induced damage (190,191).
UCP4 and UCP5 (also called brain mitochondrial carrier protein 1), which have 30% homology to UCP1 (192), are primarily expressed in the central nervous system of mammals (193,194). Although UCP4 and UCP5 are more widely distributed in the brain than UCP2, less is known about their function. UCP4 was first detected in the brain, but it has recently been found in adipocytes (195). In addition, UCP4 also plays a predominant role in insect mitochondria (196). On the other hand, UCP5, which is not limited to the brain, is also expressed in the testis, uterus, kidney, lung, stomach, liver and heart (149). It has been demonstrated that neuronal UCP4 and UCP5 share similar conformational and proton transport activities with UCP1-UCP3 (149,197). Although UCP4 and UCP5 may play an unconfirmed role in the neural system, their function for reducing oxidative stress is clear (195,198). Hoang et al (149) speculated that UCP4 acts in a neuroprotective role during early neuronal development, while UCP2 and UCP5 provided the protective function of restricting ROS production in developed neurons and other tissues, based on the observation that UCP2 and UCP5 displayed higher proton transport rates than UCP4. Oxidative stress has been proven to be involved in both neurodegenerative diseases and aging, so the UCP-dependent reduction of ROS in the nervous system has the potential to be neuroprotective in diseases such as Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis (199,200). Certain evidence indicates that protein-protein interactions exist between UCP4 and CII: UCP4-overexpressing neuroblastoma cells increase ATP synthesis via increasing the succinate-induced respiration mediated by CII (201). Pfeiffer et al (202) demonstrated that the Caenorhabditis elegans UCP4 plays a novel role in the regulation of CII by controlling succinate transport into mitochondria. UCP4 was also deemed to regulate calcium homeostasis in neuronal cells (203).
UCPs and diseases
Regardless, as an inner mitochondrial membrane protein, UCP1 mainly plays a role in the maintenance of body temperature in a cold environment through non-shivering thermogenesis and UCP2-5 can protect cells from oxidative stress by reducing the mitochondrial membrane potential via mediating uncoupling. Due to their wide distribution, UCPs have different physiological significance in specific tissues. Therefore, abnormal changes in UCPs in each tissue will lead to tissue-specific diseases (Fig. 2).
The ubiquitous UCP2 is associated with a number of metabolic diseases, such as diabetes, obesity, cardiovascular disease and even cancer, which has created immense interest in exploring the relationship between UCP2 and these diseases. Since UCP2 can regulate fatty acid and lipid metabolism, a number of studies have confirmed that UCP2 overexpression is associated with diet-induced obesity (204-206). UCP2 is highly expressed in pancreatic β-cells and has a negative regulatory effect on insulin secretion. Robson-Doucette et al (207) in 2011 demonstrated that the overexpression of UCP2 could reduce the secretion of glucose-induced insulin and subsequently induce type II diabetes (T2DM). Moreover, UCP2 knockout mice exhibited hyperinsulinemia and hypo-glycaemia (208). Briefly, glucose is metabolized through the ETC to increase ATP production, which leads to the release of insulin and the production of ROS. Chronic elevated glucose status can lead to the excessive expression of UCP2 to reduce the overproduction of ROS, resulting in reduced ATP production, reduced insulin secretion and, eventually, progression to diabetes (209,210). Chronic inflammation, including atherosclerosis, hypertension, diabetic vasculopathy and ischemia-reperfusion injury, is accompanied by excessive ROS production, which means that UCP2 can play a protective role in these diseases by reducing oxidative stress. The signaling pathways, such as nuclear factor (NF)-κB and p53, that can lead to cellular senescence, inflammation, and irreversible vasoconstriction can be inhibited by reducing ROS production. A study showed that the protein levels of UCP2 were significantly higher in human tumor tissues from the head and neck, skin, prostate and pancreas (211). Although the role of UCP2 in tumors is intuitive, the regulatory effects of UCP2 on cellular glucose and lipid metabolism, as well as the regulation of cellular oxidative stress, may be related to the development of tumors. Several studies have confirmed that UCP2 overexpression provides protection for tumor cells and leads to chemoresistance (212-214).
UCP3 has been verified to be associated with exercise intolerance in chronic obstructive pulmonary disease (COPD) patients. COPD patients have exhibited impairment in the form of exercise intolerance, which was linked to increased levels of intramuscular lipid peroxidation products (114,215). Given the fiber-type-specific expression of UCP3, researchers have examined UCP3 levels in muscle biopsies from COPD patients and found that UCP3 content was reduced in COPD (216). It can be speculated that low muscle UCP3 levels contribute to impaired exercise tolerance in COPD patients based on the function of fatty acid anion transportation. In addition, a number of studies have demonstrated that the accumulation of lipid peroxide damage, resulting from decreased UCP3 in skeletal muscle, leads to excessive oxidative stress and is a crucial aspect in the development and progression of obesity and T2DM (191,217,218).
UCP4, UCP5, together with UCP2, are expressed in the nervous system and are implicated in several neurological disorders, such as schizophrenia (SCZ), Alzheimer's disease (AD), and Parkinson's disease (PD). Various studies link UCP2 with neurodegeneration and aging (123,124,219). The three UCPs exert neuroprotective effects mainly through the alleviation of oxidative stress. The results from selected single nucleotide polymorphism markers within the neuronal UCPs showed that UCP2 and UCP4 are important in the genetic etiology of SCZ (219). Surprisingly, despite the downregulation of UCP2 mRNA levels in SCZ patients, Gigante et al (220) found that there were no differences in UCP2 protein between patients and controls. Future studies will be necessary to clarify whether the mechanism of UCP2 is protective and opposes SCZ progression. Furthermore, in the brains of AD patients, the expression levels of UCP2, 4, and 5 were significantly reduced, which limited the activation of cytoprotective mechanisms to slow the progress of AD (124). UCP5 and, especially, UCP4 are linked to PD. UCP4 is regulated by the oxidized DJ-1 and partially via the NF-κB pathway and can protect against oxidative stress and stabilize Ca2+ homeostasis in PD, as demonstrated by Ramsden et al (148) in 2012. Drugs that induce neuronal UCP expression might represent another effective strategy to ameliorate PD.
5. Concluding remarks and perspective
In conclusion, the ETC is the core component of mitochondria. The OXPHOS process in the ETC, coupled with the generation of ATP, also results in ROS production. As a double-edged sword, ROS can play a role in signaling pathways, but ROS overproduction can cause cellular damage. The ROS produced by CIII is not only released into the matrix but also released into the IMS. The ROS released into the IMS can be converted to H2O2 in a reaction catalyzed by SOD1, and the H2O2 may diffuse out of the mitochondria and play an important role in physiological and pathological pathways. Therefore, the artificial regulation of ROS at the CIII site (site IIIQo) may be of great significance. Although the precise mechanism of ROS production is still unclear, the use of specific ROS inhibitors to reduce the excessive production of ROS under pathological conditions has ameliorated oxidative stress-mediated diseases. UCP-mediated proton leak is a positive feedback mechanism for the protection of cells against oxidative stress due to the rapid production of ATP. The proper activation of UCPs can reduce the production of cellular ROS without causing a decrease in ATP; however, when the expression of UCPs becomes too high or too low or the UCP genes are mutated, pathological effects that are involved in various diseases can occur. UCP1 mediates the inducible proton conductance that is responsible for non-shivering thermogenesis in BAT, a critical response to prolonged cold exposure. UCP2 is involved in a variety of diseases, such as diabetes, obesity and cardiovascular disease. In addition, UCP2, UCP4 and UCP5 play an important role in neuroprotection and are associated with neurological diseases such as SCZ, AD and PD. Drugs targeting UCP expression and activity might represent as an effective strategy to ameliorate these diseases. However, the detailed mechanisms of the role of UCPs and the regulation of UCP expression under normal and stressful situations warrant further exploration.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81571844).
Availability of data and materials
Not applicable.
Authors' contributions
ZBY and LZ conceived the review and analyzed the relevant literature. RZZ and SJ collected and reviewed the literature related to the topic of this manuscript and drafted the main part of this manuscript. RZZ, LZ and ZBY critically revised the manuscript. RZZ and SJ produced the figures. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
References
|
Mitchell P: Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature. 191:144–148. 1961. View Article : Google Scholar : PubMed/NCBI | |
|
Guo R, Zong S, Wu M, Gu J and Yang M: Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell. 170:1247–1257.e1212. 2017. View Article : Google Scholar | |
|
Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S and Jap BK: Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 281:64–71. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Sazanov LA and Hinchliffe P: Structure of the hydrophilic domain of respiratory complex I from thermus thermophilus. Science. 311:1430–1436. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Efremov RG and Sazanov LA: Structure of the membrane domain of respiratory complex I. Nature. 476:414–420. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Jones AJ, Blaza JN, Varghese F and Hirst J: Respiratory complex I in Bos taurus and paracoccus denitrificans pumps four protons across the membrane for every NADH Oxidized. J Biol Chem. 292:4987–4995. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J and Walker JE: Bovine complex I is a complex of 45 different subunits. J Biol Chem. 281:32724–32727. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Vinothkumar KR, Zhu J and Hirst J: Architecture of mammalian respiratory complex I. Nature. 515:80–84. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Ohnishi ST, Shinzawa-Itoh K, Ohta K, Yoshikawa S and Ohnishi T: New insights into the superoxide generation sites in bovine heart NADH-ubiquinone oxidoreductase (Complex I): The significance of protein-associated ubiquinone and the dynamic shifting of generation sites between semiflavin and semiquinone radicals. Biochim Biophys Acta. 1797:1901–1909. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Gai Z, Matsuno A, Kato K, Kato S, Khan MRI, Shimizu T, Yoshioka T, Kato Y, Kishimura H, Kanno G, et al: Crystal structure of the 3.8-MDa respiratory supermolecule hemocyanin at 3.0 A resolution. Structure. 23:2204–2212. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Hunte C, Zickermann V and Brandt U: Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science. 329:448–451. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Formosa LE, Dibley MG, Stroud DA and Ryan MT: Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin Cell Dev Biol. 76:154–162. 2018. View Article : Google Scholar | |
|
Berrisford JM and Sazanov LA: Structural basis for the mechanism of respiratory complex I. J Biol Chem. 284:29773–29783. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Tan P, Feng Z, Zhang L, Hou T and Li Y: The mechanism of proton translocation in respiratory complex I from molecular dynamics. J Recept Signal Transduct Res. 35:170–179. 2015. View Article : Google Scholar | |
|
Wikstrom M and Hummer G: Stoichiometry of proton translocation by respiratory complex I and its mechanistic implications. Proc Natl Acad Sci USA. 109:4431–4436. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Chance B and Williams GR: Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J Biol Chem. 217:429–438. 1955.PubMed/NCBI | |
|
Stoner CD: Determination of the P/2e-stoichiometries at the individual coupling sites in mitochondrial oxidative phosphorylation. Evidence for maximum values of 1.0, 0.5, and 1.0 at sites 1, 2, and 3. J Biol Chem. 262:10445–10453. 1987.PubMed/NCBI | |
|
Ohnishi T: Structural biology: Piston drives a proton pump. Nature. 465:428–429. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Cecchini G: Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 72:77–109. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M and Rao Z: Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 121:1043–1057. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Bezawork-Geleta A, Rohlena J, Dong L, Pacak K and Neuzil J: Mitochondrial complex II: At the crossroads. Trends Biochem Sci. 42:312–325. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Iverson TM: Catalytic mechanisms of complex II enzymes: A structural perspective. Biochim Biophys Acta. 1827:648–657. 2013. View Article : Google Scholar | |
|
Schagger H, Link TA, Engel WD and von Jagow G: Isolation of the eleven protein subunits of the bc1 complex from beef heart. Methods Enzymol. 126:224–237. 1986. View Article : Google Scholar : PubMed/NCBI | |
|
Yang XH and Trumpower BL: Purification of a three-subunit ubiquinol-cytochrome c oxidoreductase complex from paracoccus denitrificans. J Biol Chem. 261:12282–12289. 1986.PubMed/NCBI | |
|
Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA and Xia D: Structural basis for the quinone reduction in the bc1 complex: A comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry. 42:9067–9080. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Mitchell P: Chemiosmotic coupling in energy transduction: A logical development of biochemical knowledge. J Bioenerg. 3:5–24. 1972. View Article : Google Scholar : PubMed/NCBI | |
|
Mitchell P: Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol. 62:327–367. 1976. View Article : Google Scholar : PubMed/NCBI | |
|
Trumpower BL: A concerted, alternating sites mechanism of ubiquinol oxidation by the dimeric cytochrome bc(1) complex. Biochim Biophys Acta. 1555:166–173. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Kadenbach B and Hüttemann M: The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion. 24:64–76. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R and Yoshikawa S: The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 272:1136–1144. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Konstantinov AA: Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 586:630–639. 2012. View Article : Google Scholar | |
|
Sharma V and Wikstrom M: The role of the K-channel and the active-site tyrosine in the catalytic mechanism of cytochrome c oxidase. Biochim Biophys Acta. 1857:1111–1115. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Varanasi L and Hosler JP: Subunit III-depleted cytochrome c oxidase provides insight into the process of proton uptake by proteins. Biochim Biophys Acta. 1817:545–551. 2012. View Article : Google Scholar : | |
|
Alnajjar KS, Hosler J and Prochaska L: Role of the N-terminus of subunit III in proton uptake in cytochrome c oxidase of Rhodobacter sphaeroides. Biochemistry. 53:496–504. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Arnold S and Kadenbach B: Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome-c oxidase. Eur J Biochem. 249:350–354. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Arnold S and Kadenbach B: The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosteri-cally. FEBS Lett. 443:105–108. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Arnold S, Goglia F and Kadenbach B: 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur J Biochem. 252:325–330. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Follmann K, Arnold S, Ferguson-Miller S and Kadenbach B: Cytochrome c oxidase from eucaryotes but not from procaryotes is allosterically inhibited by ATP. Biochem Mol Biol Int. 45:1047–1055. 1998.PubMed/NCBI | |
|
Shimada S, Shinzawa-Itoh K, Baba J, Aoe S, Shimada A, Yamashita E, Kang J, Tateno M, Yoshikawa S and Tsukihara T: Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J. 36:291–300. 2017. View Article : Google Scholar | |
|
Wikstrom MK: Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 266:271–273. 1977. View Article : Google Scholar : PubMed/NCBI | |
|
Jonckheere AI, Smeitink JA and Rodenburg RJ: Mitochondrial ATP synthase: Architecture, function and pathology. J Inherit Metab Dis. 35:211–225. 2012. View Article : Google Scholar : | |
|
Dickson VK, Silvester JA, Fearnley IM, Leslie AG and Walker JE: On the structure of the stator of the mitochondrial ATP synthase. EMBO J. 25:2911–2918. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Watt IN, Montgomery MG, Runswick MJ, Leslie AG and Walker JE: Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA. 107:16823–16827. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Pecina P, Nůsková H, Karbanová V, Kaplanová V, Mráček T and Houštěk J: Role of the mitochondrial ATP synthase central stalk subunits γ and δ in the activity and assembly of the mammalian enzyme. Biochim Biophys Acta Bioenerg. 1859:374–381. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Guo R, Gu J, Zong S, Wu M and Yang M: Structure and mechanism of mitochondrial electron transport chain. Biomed J. 41:9–20. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M and Sazanov LA: Atomic structure of the entire mammalian mitochondrial complex I. Nature. 538:406–410. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W and Meier T: Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol Cell. 63:445–456. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Turrens JF: Mitochondrial formation of reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Cadenas E and Davies KJ: Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 29:222–230. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Brand MD: Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 100:14–31. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF and Vercesi AE: Mitochondria and reactive oxygen species. Free Radic Biol Med. 47:333–343. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Brand MD: The sites and topology of mitochondrial superoxide production. Exp Gerontol. 45:466–472. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Hernansanz-Agustin P, Ramos E, Navarro E, Parada E, Sánchez-López N, Peláez-Aguado L, Cabrera-García JD, Tello D, Buendia I, Marina A, et al: Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 12:1040–1051. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hoekstra AS and Bayley JP: The role of complex II in disease. Biochim Biophys Acta. 1827:543–551. 2013. View Article : Google Scholar | |
|
Cecchini G: Respiratory complex II: Role in cellular physiology and disease. Biochim Biophys Acta. 1827:541–542. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA and Brand MD: Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 287:27255–27264. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Ackrell BA, Kearney EB and Singer TP: Mammalian succinate dehydrogenase. Methods Enzymol. 53:466–483. 1978. View Article : Google Scholar : PubMed/NCBI | |
|
Turrens JF, Alexandre A and Lehninger AL: Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 237:408–414. 1985. View Article : Google Scholar : PubMed/NCBI | |
|
Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, Walter P and Yaffee M: Oxidants in mitochondria: From physiology to diseases. Biochim Biophys Acta. 1271:67–74. 1995. View Article : Google Scholar : PubMed/NCBI | |
|
Muller FL, Liu Y and Van Remmen H: Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 279:49064–49073. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
D'Autreaux B and Toledano MB: ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Bedard K and Krause KH: The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Treberg JR, Quinlan CL and Brand MD: Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production-a correction using glutathione depletion. FEBS J. 277:2766–2778. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Quinlan CL, Gerencser AA, Treberg JR and Brand MD: The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem. 286:31361–31372. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Erecinska M and Wilson DF: The effect of antimycin A on cytochromes b561, b566, and their relationship to ubiquinone and the iron-sulfer centers S-1 (+N-2) and S-3. Arch Biochem Biophys. 174:143–157. 1976. View Article : Google Scholar : PubMed/NCBI | |
|
Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, et al: Suppressors of superoxide production from mitochondrial complex III. Nat Chem Biol. 11:834–836. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Muramoto K, Ohta K, Shinzawa-Itoh K, Kanda K, Taniguchi M, Nabekura H, Yamashita E, Tsukihara T and Yoshikawa S: Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc Natl Acad Sci USA. 107:7740–7745. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
De Giusti VC, Caldiz CI, Ennis IL, Perez NG, Cingolani HE and Aiello EA: Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS). Front Physiol. 4:1262013. View Article : Google Scholar : PubMed/NCBI | |
|
Diebold L and Chandel NS: Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 100:86–93. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Kaminskyy VO and Zhivotovsky B: Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. 21:86–102. 2014. View Article : Google Scholar | |
|
Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, Budinger GR and Chandel NS: Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 46:1386–1391. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Betzen C, White R, Zehendner CM, Pietrowski E, Bender B, Luhmann HJ and Kuhlmann CR: Oxidative stress upregulates the NMDA receptor on cerebrovascular endothelium. Free Radic Biol Med. 47:1212–1220. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Huddleston AT, Tang W, Takeshima H, Hamilton SL and Klann E: Superoxide-induced potentiation in the hippocampus requires activation of ryanodine receptor type 3 and ERK. J Neurophysiol. 99:1565–1571. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Hidalgo C and Arias-Cavieres A: Calcium, reactive oxygen species, and synaptic plasticity. Physiology (Bethesda). 31:201–215. 2016. | |
|
Shetty PK, Huang FL and Huang KP: Ischemia-elicited oxidative modulation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 283:5389–5401. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Kemmerling U, Munoz P, Muller M, Sánchez G, Aylwin ML, Klann E, Carrasco MA and Hidalgo C: Calcium release by ryanodine receptors mediates hydrogen peroxide-induced activation of ERK and CREB phosphorylation in N2a cells and hippocampal neurons. Cell Calcium. 41:491–502. 2007. View Article : Google Scholar | |
|
Massaad CA and Klann E: Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 14:2013–2054. 2011. View Article : Google Scholar : | |
|
Beckhauser TF, Francis-Oliveira J and De Pasquale R: Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J Exp Neurosci. 10(Suppl 1): S23–S48. 2016. | |
|
Gasperini RJ, Pavez M, Thompson AC, Mitchell CB, Hardy H, Young KM, Chilton JK and Foa L: How does calcium interact with the cytoskeleton to regulate growth cone motility during axon pathfinding? Mol Cell Neurosci. 84:29–35. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Oswald MCW, Garnham N, Sweeney ST and Landgraf M: Regulation of neuronal development and function by ROS. FEBS Lett. 592:679–691. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Hongpaisan J, Winters CA and Andrews SB: Calcium-dependent mitochondrial superoxide modulates nuclear CREB phosphorylation in hippocampal neurons. Mol Cell Neurosci. 24:1103–1115. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Orrenius S, Gogvadze V and Zhivotovsky B: Mitochondrial oxidative stress: Implications for cell death. Annu Rev Pharmacol Toxicol. 47:143–183. 2007. View Article : Google Scholar | |
|
Cingolani HE, Perez NG, Aiello EA, Ennis IL, Garciarena CD, Villa-Abrille MC, Dulce RA, Caldiz CI, Yeves AM, Correa MV, et al: Early signals after stretch leading to cardiac hypertrophy. Key role of NHE-1. Front Biosci. 13:7096–7114. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV and Mattiazzi A: Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 105:1204–1212. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Wang H and Patterson C: Roles of reactive oxygen species in physiology and pathology. John Wiley & Sons, Inc; Hoboken, NJ: pp. 379–392. 2015 | |
|
Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, Hoek TL and Schumacker PT: Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J Biol Chem. 282:19133–19143. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH and Rojanasakul Y: The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 180:3072–3080. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Circu ML and Aw TY: Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Deng Y, Ren X, Yang L, Lin Y and Wu X: A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 115:61–70. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Hattori K, Naguro I, Runchel C and Ichijo H: The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 7:92009. View Article : Google Scholar : PubMed/NCBI | |
|
Sinha K, Das J, Pal PB and Sil PC: Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Shen C, Cai GQ, Peng JP and Chen XD: Autophagy protects chondrocytes from glucocorticoids-induced apoptosis via ROS/Akt/FOXO3 signaling. Osteoarthritis Cartilage. 23:2279–2287. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Shah SZA, Zhao D, Hussain T, Sabir N, Mangi MH and Yang L: p62-Keap1-NRF2-ARE pathway: A contentious player for selective targeting of autophagy, oxidative stress and mitochondrial dysfunction in prion diseases. Front Mol Neurosci. 11:3102018. View Article : Google Scholar : PubMed/NCBI | |
|
Wu H, Huang S, Chen Z, Liu W, Zhou X and Zhang D: Hypoxia-induced autophagy contributes to the invasion of salivary adenoid cystic carcinoma through the HIF-1α/BNIP3 signaling pathway. Mol Med Rep. 12:6467–6474. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Bensaad K, Cheung EC and Vousden KH: Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 28:3015–3026. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Gurusamy N and Das DK: Autophagy, redox signaling, and ventricular remodeling. Antioxid Redox Signal. 11:1975–1988. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Li R, Zhou P, Guo Y, Lee JS and Zhou B: Tris (1, 3-dichloro-2-propyl) phosphate induces apoptosis and autophagy in SH-SY5Y cells: Involvement of ROS-mediated AMPK/mTOR/ULK1 pathways. Food Chem Toxicol. 100:183–196. 2017. View Article : Google Scholar | |
|
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 15:135–147. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Schenk B and Fulda S: Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene. 34:5796–5806. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Morgan MJ and Liu ZG: Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol Cells. 30:1–12. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Ying Y, Kim J, Westphal SN, Long KE and Padanilam BJ: Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 25:2707–2716. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC and Salter RD: Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 191:5230–5238. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou R, Tardivel A, Thorens B, Choi I and Tschopp J: Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 11:136–140. 2010. View Article : Google Scholar | |
|
Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J and Khavari PA: Mek1/2 MAPK kinases are essential for mammalian development, homeostasis, and raf-induced hyperplasia. Dev Cell. 12:615–629. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Hulsmans M, Van Dooren E and Holvoet P: Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr Atheroscler Rep. 14:264–276. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Kattoor AJ, Pothineni NVK, Palagiri D and Mehta JL: Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 19:422017. View Article : Google Scholar : PubMed/NCBI | |
|
Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM and Touyz RM: Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol. 31:631–641. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K and Huttemann M: Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 47:9–23. 2013. View Article : Google Scholar : | |
|
Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T and Murphy MP: A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23:254–263. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Lorenzo O, Ramirez E, Picatoste B, Egido J and Tunon J: Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediators Inflamm. 2013:4619672013. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao MX, Zhou B, Ling L, Xiong XQ, Zhang F, Chen Q, Li YH, Kang YM and Zhu GQ: Salusin-beta contributes to oxidative stress and inflammation in diabetic cardiomyopathy. Cell Death Dis. 8:e26902017. View Article : Google Scholar | |
|
Jaitovich A and Jourd'Heuil D: A brief overview of nitric oxide and reactive oxygen species signaling in hypoxia-induced pulmonary hypertension. Adv Exp Med Biol. 967:71–81. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Fulton DJR, Li X, Bordan Z, Haigh S, Bentley A, Chen F and Barman SA: Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants (Basel). 6:E542017. View Article : Google Scholar | |
|
Allaire J, Maltais F, LeBlanc P, Simard PM, Whittom F, Doyon JF, Simard C and Jobin J: Lipofuscin accumulation in the vastus lateralis muscle in patients with chronic obstructive pulmonary disease. Muscle Nerve. 25:383–389. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL and Bast A: Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 154:813–816. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA and Barnes PJ: Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 162:1175–1177. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Sabharwal SS and Schumacker PT: Mitochondrial ROS in cancer: Initiators, amplifiers or an achilles' heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H and Lleonart ME: Oxidative stress and cancer: An overview. Ageing Res Rev. 12:376–390. 2013. View Article : Google Scholar | |
|
Ahmad W, Ijaz B, Shabbiri K, Ahmed F and Rehman S: Oxidative toxicity in diabetes and Alzheimer's disease: Mechanisms behind ROS/RNS generation. J Biomed Sci. 24:762017. View Article : Google Scholar | |
|
Rehman K and Akash MSH: Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: How are they interlinked? J Cell Biochem. 118:3577–3585. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Spahis S, Delvin E, Borys JM and Levy E: Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid Redox Signal. 26:519–541. 2017. View Article : Google Scholar | |
|
Rolo AP, Teodoro JS and Palmeira CM: Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 52:59–69. 2012. View Article : Google Scholar | |
|
de la Monte SM and Wands JR: Molecular indices of oxidative stress and mitochondrial dysfunction occur early and oftenprogresswith severity of Alzheimer's disease. J Alzheimers Dis. 9:167–181. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Dias V, Junn E and Mouradian MM: The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 3:461–491. 2013.PubMed/NCBI | |
|
Nagano T, Mizuno M, Morita K and Nawa H: Pathological implications of oxidative stress in patients and animal models with schizophrenia: The role of epidermal growth factor receptor signaling. Curr Top Behav Neurosci. 29:429–446. 2016. View Article : Google Scholar | |
|
Mahadik SP, Pillai A, Joshi S and Foster A: Prevention of oxidative stress-mediated neuropathology and improved clinical outcome by adjunctive use of a combination of antioxidants and omega-3 fatty acids in schizophrenia. Int Rev Psychiatry. 18:119–131. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Kwon DN, Park WJ, Choi YJ, Gurunathan S and Kim JH: Oxidative stress and ROS metabolism via down-regulation of sirtuin 3 expression in Cmahnull mice affect hearing loss. Aging (Albany NY). 7:579–594. 2015. View Article : Google Scholar | |
|
Kamogashira T, Fujimoto C and Yamasoba T: Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed Res Int. 2015:6172072015. View Article : Google Scholar : PubMed/NCBI | |
|
Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM and Suburo AM: Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: Implications for age-related macular degeneration. Redox Biol. 7:78–87. 2016. View Article : Google Scholar : | |
|
Nita M and Grzybowski A: The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016:31647342016. View Article : Google Scholar : PubMed/NCBI | |
|
Ghosh S, Sulistyoningrum DC, Glier MB, Verchere CB and Devlin AM: Altered glutathione homeostasis in heart augments cardiac lipotoxicity associated with diet-induced obesity in mice. J Biol Chem. 286:42483–42493. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Mandas A, Iorio EL, Congiu MG, Balestrieri C, Mereu A, Cau D, Dessì S and Curreli N: Oxidative imbalance in HIV-1 infected patients treated with antiretroviral therapy. J Biomed Biotechnol. 2009:7495752009. View Article : Google Scholar : PubMed/NCBI | |
|
Shin DH, Martinez SS, Parsons M, Jayaweera DT, Campa A and Baum MK: Relationship of oxidative stress with HIV disease progression in HIV/HCV Co-infected and HIV mono-infected adults in miami. Int J Biosci Biochem Bioinforma. 2:217–223. 2012.PubMed/NCBI | |
|
Haycock JW, MacNeil S, Jones P, Harris JB and Mantle D: Oxidative damage to muscle protein in Duchenne muscular dystrophy. Neuroreport. 8:357–361. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Austin L, de Niese M, McGregor A, Arthur H, Gurusinghe A and Gould MK: Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscul Disord. 2:27–33. 1992. View Article : Google Scholar : PubMed/NCBI | |
|
Lee G, Kim HJ and Kim HM: RhoA-JNK regulates the E-cadherin junctions of human gingival epithelial cells. J Dent Res. 95:284–291. 2016. View Article : Google Scholar | |
|
Nobes CD, Brown GC, Olive PN and Brand MD: Nonohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J Biol Chem. 265:12903–12909. 1990.PubMed/NCBI | |
|
Brand MD, Turner N, Ocloo A, Else PL and Hulbert AJ: Proton conductance and fatty acyl composition of liver mitochondria correlates with body mass in birds. Biochem J. 376:741–748. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Porter RK and Brand MD: Body mass dependence of H+ leak in mitochondria and its relevance to metabolic rate. Nature. 362:628–630. 1993. View Article : Google Scholar : PubMed/NCBI | |
|
Brookes PS, Rolfe DF and Brand MD: The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: Comparison with isolated mitochondria. J Membr Biol. 155:167–174. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS and Cornwall EJ: The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 392:353–362. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Parker N, Crichton PG, Vidal-Puig AJ and Brand MD: Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr. 41:335–342. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Shabalina IG, Ost M, Petrovic N, Vrbacky M, Nedergaard J and Cannon B: Uncoupling protein-1 is not leaky. Biochim Biophys Acta. 1797:773–784. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Roussel D, Harding M, Runswick MJ, Walker JE and Brand MD: Does any yeast mitochondrial carrier have a native uncoupling protein function? J Bioenerg Biomembr. 34:165–176. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Krauss S, Zhang CY and Lowell BB: The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 6:248–261. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Huang SG and Klingenberg M: Fluorescent nucleotide derivatives as specific probes for the uncoupling protein: Thermodynamics and kinetics of binding and the control by pH. Biochemistry. 34:349–360. 1995. View Article : Google Scholar : PubMed/NCBI | |
|
Xia C, Liu JZ and Xu Y: Effects of GDP on the activity and expression of mitochondrial uncoupling proteins in rat brain in vitro. Sheng Li Xue Bao. 60:492–496. 2008.In Chinese. PubMed/NCBI | |
|
Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH and Ho SL: Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): Structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2:468–478. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Hoang T, Smith MD and Jelokhani-Niaraki M: Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry. 51:4004–4014. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Andrews ZB, Diano S and Horvath TL: Mitochondrial uncoupling proteins in the cns: In support of function and survival. Nat Rev Neurosci. 6:829–840. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Nicholls DG and Locke RM: Thermogenic mechanisms in brown fat. Physiol Rev. 64:1–64. 1984. View Article : Google Scholar : PubMed/NCBI | |
|
Shabalina IG, Petrovic N, de Jong JM, Kalinovich AV, Cannon B and Nedergaard J: UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 5:1196–1203. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Golozoubova V, Hohtola E, Matthias A, Jacobsson A, Cannon B and Nedergaard J: Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 15:2048–2050. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME and Kozak LP: Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 387:90–94. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Divakaruni AS and Brand MD: The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda). 26:192–205. 2011. | |
|
Granneman JG, Burnazi M, Zhu Z and Schwamb LA: White adipose tissue contributes to UCP1-independent thermogenesis. Am J Physiol Endocrinol Metab. 285:E1230–E1236. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Adams AE, Kelly OM and Porter RK: Absence of mitochondrial uncoupling protein 1 affects apoptosis in thymocytes, thymocyte/T-cell profile and peripheral T-cell number. Biochim Biophys Acta. 1797:807–816. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Adams AE, Hanrahan O, Nolan DN, Voorheis HP, Fallon P and Porter RK: Images of mitochondrial UCP 1 in mouse thymocytes using confocal microscopy. Biochim Biophys Acta. 1777:115–117. 2008. View Article : Google Scholar | |
|
Adams AE, Carroll AM, Fallon PG and Porter RK: Mitochondrial uncoupling protein 1 expression in thymocytes. Biochim Biophys Acta. 1777:772–776. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Sale MM, Hsu FC, Palmer ND, Gordon CJ, Keene KL, Borgerink HM, Sharma AJ, Bergman RN, Taylor KD, Saad MF and Norris JM: The uncoupling protein 1 gene, UCP1, is expressed in mammalian islet cells and associated with acute insulin response to glucose in african american families from the IRAS family study. BMC Endocr Disord. 7:12007. View Article : Google Scholar : PubMed/NCBI | |
|
Shabalina IG, Jacobsson A, Cannon B and Nedergaard J: Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 279:38236–38248. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Breen EP, Gouin SG, Murphy AF, Haines LR, Jackson AM, Pearson TW, Murphy PV and Porter RK: On the mechanism of mitochondrial uncoupling protein 1 function. J Biol Chem. 281:2114–2119. 2006. View Article : Google Scholar | |
|
Rial E, Aguirregoitia E, Jimenez-Jimenez J and Ledesma A: Alkylsulfonates activate the uncoupling protein UCP1: Implications for the transport mechanism. Biochim Biophys Acta. 1608:122–130. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Klingenberg M and Huang SG: Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta. 1415:271–296. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Garlid KD, Jaburek M and Jezek P: The mechanism of proton transport mediated by mitochondrial uncoupling proteins. FEBS Lett. 438:10–14. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Garlid KD, Orosz DE, Modriansky M, Vassanelli S and Jezek P: On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J Biol Chem. 271:2615–2620. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Winkler E and Klingenberg M: Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J Biol Chem. 269:2508–2515. 1994.PubMed/NCBI | |
|
Skulachev VP: Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 294:158–162. 1991. View Article : Google Scholar : PubMed/NCBI | |
|
Klingenberg M and Winkler E: The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 4:3087–3092. 1985. View Article : Google Scholar : PubMed/NCBI | |
|
Azzu V and Brand MD: The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. 35:298–307. 2010. View Article : Google Scholar | |
|
Ricquier D and Bouillaud F: The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 345(Pt 2): 161–179. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Friederich M, Fasching A, Hansell P, Nordquist L and Palm F: Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 1777:935–940. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Flachs P, Sponarova J, Kopecky P, Horvath O, Sediva A, Nibbelink M, Casteilla L, Medrikova D, Neckar J, Kolar F and Kopecky J: Mitochondrial uncoupling protein 2 gene transcript levels are elevated in maturating erythroid cells. FEBS Lett. 581:1093–1097. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Affourtit C and Brand MD: On the role of uncoupling protein-2 in pancreatic beta cells. Biochim Biophys Acta. 1777:973–979. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Cadenas S: Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. 1859:940–950. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Teshima Y, Akao M, Jones SP and Marban E: Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res. 93:192–200. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Vidal-Puig A, Solanes G, Grujic D, Flier JS and Lowell BB: UCP3: An uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun. 235:79–82. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Aguirre E and Cadenas S: GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart. Biochim Biophys Acta. 1797:1716–1726. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Kelly OM and Porter RK: Absence of mitochondrial uncoupling protein 3: Effect on thymus and spleen in the fed and fasted mice. Biochim Biophys Acta. 1807:1064–1074. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Mori S, Yoshizuka N, Takizawa M, Takema Y, Murase T, Tokimitsu I and Saito M: Expression of uncoupling proteins in human skin and skin-derived cells. J Invest Dermatol. 128:1894–1900. 2008. View Article : Google Scholar | |
|
Harper ME and Himms-Hagen J: Mitochondrial efficiency: Lessons learned from transgenic mice. Biochim Biophys Acta. 1504:159–172. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L and Casteilla L: A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 11:809–815. 1997. View Article : Google Scholar : PubMed/NCBI | |
|
Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J and Wieckowski MR: Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 810:183–205. 2012. View Article : Google Scholar | |
|
Pi J, Bai Y, Daniel KW, Liu D, Lyght O, Edelstein D, Brownlee M, Corkey BE and Collins S: Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology. 150:3040–3048. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, Szczepanik A, Wade J, Mootha V, Cortright R, et al: Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 275:16258–16266. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Brand MD, Pamplona R, Portero-Otin M, Requena JR, Roebuck SJ, Buckingham JA, Clapham JC and Cadenas S: Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem J. 368:597–603. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Brand MD: Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 35:811–820. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Dlaskova A, Clarke KJ and Porter RK: The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim Biophys Acta. 1797:1470–1476. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Shabalina IG, Vrbacky M, Pecinova A, Kalinovich AV, Drahota Z, Houštěk J, Mráček T, Cannon B and Nedergaard J: ROS production in brown adipose tissue mitochondria: The question of UCP1-dependence. Biochim Biophys Acta. 1837:2017–2030. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Skulachev VP: Anion carriers in fatty acid-mediated physiological uncoupling. J Bioenerg Biomembr. 31:431–445. 1999. View Article : Google Scholar | |
|
Schrauwen P and Hesselink MK: The role of uncoupling protein 3 in fatty acid metabolism: Protection against lipotoxicity? Proc Nutr Soc. 63:287–292. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Bouillaud F, Couplan E, Pecqueur C and Ricquier D: Homologues of the uncoupling protein from brown adipose tissue (UCP1): UCP2, UCP3, BMCP1 and UCP4. Biochim Biophys Acta. 1504:107–119. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Mao W, Yu XX, Zhong A, Li W, Brush J, Sherwood SW, Adams SH and Pan G: UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 443:326–330. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Sanchis D, Fleury C, Chomiki N, Goubern M, Huang Q, Neverova M, Grégoire F, Easlick J, Raimbault S, Lévi-Meyrueis C, et al: BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J Biol Chem. 273:34611–34615. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang M, Wang B, Ni YH, Liu F, Fei L, Pan XQ, Guo M, Chen RH and Guo XR: Overexpression of uncoupling protein 4 promotes proliferation and inhibits apoptosis and differentiation of preadipocytes. Life Sci. 79:1428–1435. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Slocinska M, Barylski J and Jarmuszkiewicz W: Uncoupling proteins of invertebrates: A review. IUBMB Life. 68:691–699. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Ivanova MV, Hoang T, McSorley FR, Krnac G, Smith MD and Jelokhani-Niaraki M: A comparative study on conformation and ligand binding of the neuronal uncoupling proteins. Biochemistry. 49:512–521. 2010. View Article : Google Scholar | |
|
Kwok KH, Ho PW, Chu AC, Ho JW, Liu HF, Yiu DC, Chan KH, Kung MH, Ramsden DB and Ho SL: Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radic Biol Med. 49:1023–1035. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Emerit J, Edeas M and Bricaire F: Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 58:39–46. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Kim-Han JS and Dugan LL: Mitochondrial uncoupling proteins in the central nervous system. Antioxid Redox Signal. 7:1173–1181. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Ho PW, Ho JW, Tse HM, So DH, Yiu DC, Liu HF, Chan KH, Kung MH, Ramsden DB and Ho SL: Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial complex II in neuroblastoma cells. PLoS One. 7:e328102012. View Article : Google Scholar : PubMed/NCBI | |
|
Pfeiffer M, Kayzer EB, Yang X, Abramson E, Kenaston MA, Lago CU, Lo HH, Sedensky MM, Lunceford A, Clarke CF, et al: Caenorhabditis elegans UCP4 protein controls complex II-mediated oxidative phosphorylation through succinate transport. J Biol Chem. 286:37712–37720. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Chan SL, Liu D, Kyriazis GA, Bagsiyao P, Ouyang X and Mattson MP: Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 281:37391–37403. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Walder K, Norman RA, Hanson RL, Schrauwen P, Neverova M, Jenkinson CP, Easlick J, Warden CH, Pecqueur C, Raimbault S, et al: Association between uncoupling protein polymorphisms (UCP2UCP3) and energy metabolism/obesity in pima indians. Hum Mol Genet. 7:1431–1435. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Pheiffer C, Jacobs C, Patel O, Ghoor S, Muller C and Louw J: Expression of UCP2 in wistar rats varies according to age and the severity of obesity. J Physiol Biochem. 72:25–32. 2016. View Article : Google Scholar | |
|
Millet L, Vidal H, Andreelli F, Larrouy D, Riou JP, Ricquier D, Laville M and Langin D: Increased uncoupling protein-2 and -3 mRNA expression during fasting in obese and lean humans. J Clin Invest. 100:2665–2670. 1997. View Article : Google Scholar | |
|
Robson-Doucette CA, Sultan S, Allister EM, Wikstrom JD, Koshkin V, Bhattacharjee A, Prentice KJ, Sereda SB, Shirihai OS and Wheeler MB: Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes. 60:2710–2719. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Gonzalez-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanné-Chantelot C, Hubert L, de Keyzer Y, de Lonlay P and Ricquier D: Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One. 3:e38502008. View Article : Google Scholar : PubMed/NCBI | |
|
Lameloise N, Muzzin P, Prentki M and Assimacopoulos-Jeannet F: Uncoupling protein 2: A possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes. 50:803–809. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Patane G, Anello M, Piro S, Vigneri R, Purrello F and Rabuazzo AM: Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes. 51:2749–2756. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Li W, Nichols K, Nathan CA and Zhao Y: Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark. 13:377–383. 2013. View Article : Google Scholar | |
|
Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR and Baffy G: The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68:2813–2819. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Pons DG, Nadal-Serrano M, Torrens-Mas M, Valle A, Oliver J and Roca P: UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic Biol Med. 86:67–77. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Kawanishi M, Fukuda T, Shimomura M, Inoue Y, Wada T, Tasaka R, Yasui T and Sumi T: Expression of UCP2 is associated with sensitivity to platinum-based chemotherapy for ovarian serous carcinoma. Oncol Lett. 15:9923–9928. 2018.PubMed/NCBI | |
|
Franssen FM, Wouters EF, Baarends EM, Akkermans MA and Schols AM: Arm mechanical efficiency and arm exercise capacity are relatively preserved in chronic obstructive pulmonary disease. Med Sci Sports Exerc. 34:1570–1576. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Gosker HR, Schrauwen P, Broekhuizen R, Hesselink MK, Moonen-Kornips E, Ward KA, Franssen FM, Wouters EF and Schols AM: Exercise training restores uncoupling protein-3 content in limb muscles of patients with chronic obstructive pulmonary disease. Am J Physiol. Endocrinol Metab. 290:E976–E981. 2006. | |
|
Patti ME and Corvera S: The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. 31:364–395. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Schrauwen P, Hesselink MK, Blaak EE, Borghouts LB, Schaart G, Saris WH and Keizer HA: Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 50:2870–2873. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Yasuno K, Ando S, Misumi S, Makino S, Kulski JK, Muratake T, Kaneko N, Amagane H, Someya T, Inoko H, et al: Synergistic association of mitochondrial uncoupling protein (UCP) genes with schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 144B:250–253. 2007. View Article : Google Scholar | |
|
Gigante AD, Andreazza AC, Lafer B, Yatham LN, Beasley CL and Young LT: Decreased mRNA expression of uncoupling protein 2, a mitochondrial proton transporter, in post-mortem prefrontal cortex from patients with bipolar disorder and schizophrenia. Neurosci Lett. 505:47–51. 2011. View Article : Google Scholar : PubMed/NCBI |