
Protein aggregation and biomolecular condensation in hypoxic environments (Review)
- Authors:
- Published online on: February 12, 2024 https://doi.org/10.3892/ijmm.2024.5357
- Article Number: 33
-
Copyright: © Li et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
1. Introduction
The concentration range of biological macromolecules such as ribonucleoproteins, polysaccharides, nucleic acids, proteins and others inside cells is 80-400 mg/ml (1). In response to such high concentrations, organisms have developed various conserved mechanisms to prevent the chaotic aggregation of proteins by allowing these proteins to form different higher-order complexes with multiple biological functions as a response to different types of environmental stress (2).
There are typically two types of higher-order assemblies: i) Stable and rigid protein-protein interactions that generate ordered, solid-like macromolecular complexes; and ii) complexes consisting of weaker and more dynamic molecules. In biology, the term 'aggregation' is commonly used to describe assemblies formed under pathological conditions, where the molecules in the aggregate are irreversibly disrupted and often considered as pathogenic factors. Aggregation represents a prominent characteristic of irreversible biological processes. By contrast, the term 'condensation' refers to reversible and dynamic molecules which can be redissolved to perform their respective functions, and their assembly is tightly monitored within the intracellular environment (3). However, these two types of higher-order protein assemblers are not completely independent. Disruptions in protein homeostasis under pressure or under pathological conditions can result in an imbalance of biomolecular condensation, ultimately leading to the uncontrolled collapse of these structures, which in turn triggers the irreversible aggregation and misfolding of protein constituents, and often leads to the transformation of aged or solidified condensates into aggregates (4,5).
Hypoxia is a prevalent environmental stressor encountered by aerobic organisms and a common property of pathological disorders such as bacterial infections, inflammation, impairment, cardiovascular disease (CVD) and cancer (6,7). Eukaryotes have developed a rapid and well-conserved hypoxia response mechanism. More specifically, hypoxia induces the production of cellular reactive oxygen species (ROS) and acidification of the cellular environment due to decreased oxygen supply (8). Several studies have examined the stress responses of mitochondria and endoplasmic reticulum (ER) under hypoxic conditions, and they showed that the protein folding process is impaired and protein homeostasis is disrupted (9,10). Kaufman et al (9) recently revealed that hypoxia-induced insolubility of specific proteins in nematodes; it was revealed that oxygen depletion and adenosine triphosphate (ATP) could disturb the intracellular equilibrium, leading to uncontrolled aggregation. However, eukaryotic cells have evolved conserved molecular chaperones and protein autophagy networks to maintain balance (6). There is also increasing evidence that uncontrolled protein homeostasis and condensate aging are involved in hypoxia-related diseases, providing a probable cause for the relationship between hypoxic stress and related diseases (11-13).
A hypoxic environment may induce an imbalance of protein homeostasis and aggregation. This imbalance can also activate the assembly of biomolecular condensates, which play crucial roles as organelles without membrane and are regulated by multiple mechanisms related to environmental stress (3). Stress granules (SGs) (14), glycolytic bodies (G-bodies) (15) and processing bodies (P-bodies) (16) contribute to cell survival under stress conditions and induce metabolic reprogramming in hypoxic environments.
In the present review, the aim was to summarize hypoxia-induced aggregate behaviors and discuss their functions and regulatory mechanisms, hoping that the information provided in the review could help us to gain better insights into the mechanisms underlying neuromedicine, altitude medicine and the tumor microenvironment.
2. Hypoxia-induced protein aggregation and regulatory responses
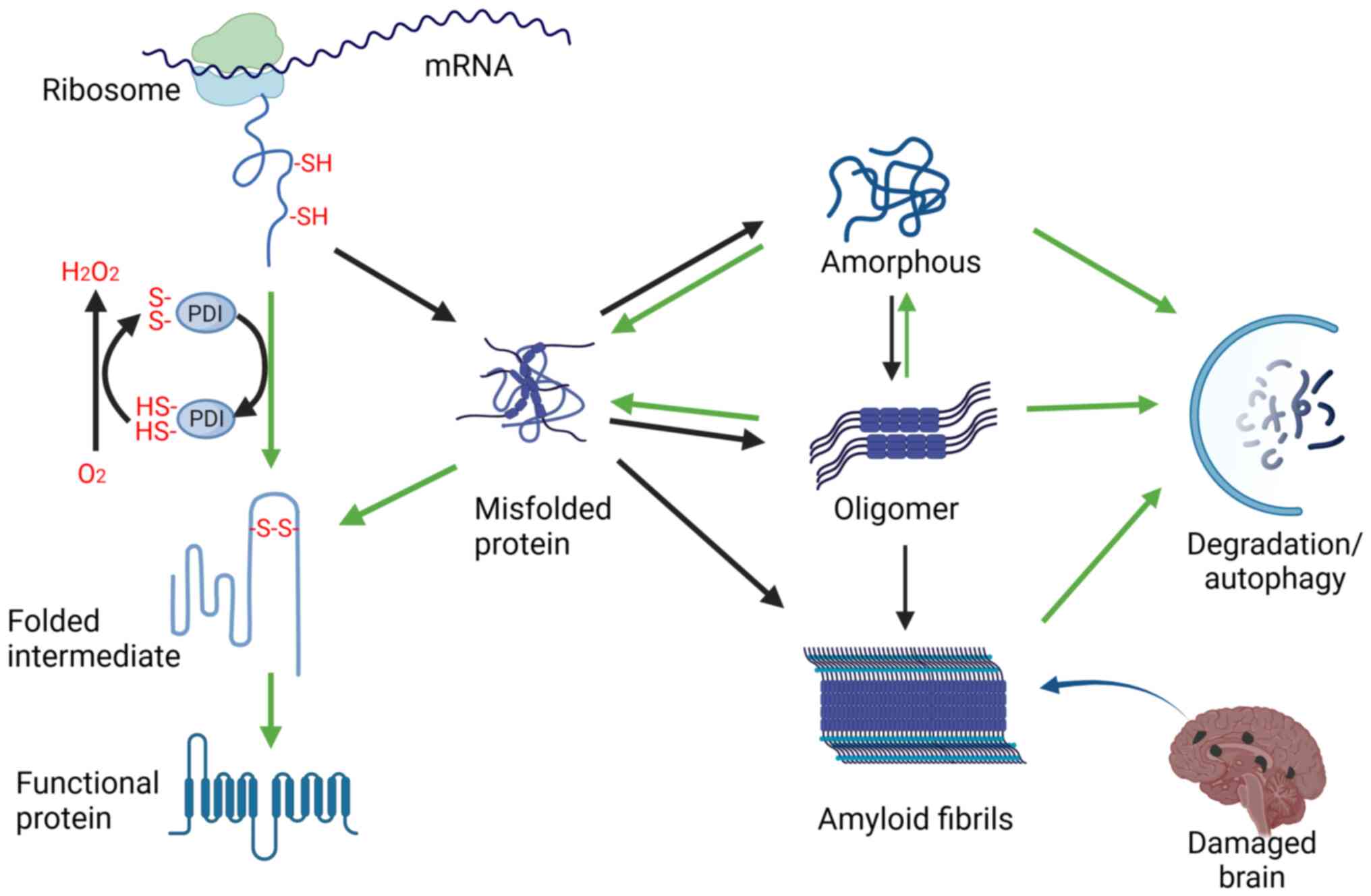
Mechanisms of hypoxia-induced unfolded/misfolded protein aggregation
Hypoxia is a common stressor for aerobic cells that can lead to cell acidification, oxidative stress, cell cycle arrest and death (17). Using transmission electron microscopy, recent studies have revealed the presence of abundant electron-dense deposits, which represent aggregates of unfolded and misfolded proteins in neurons exposed to ischemic-hypoxic brain injury (18,19). During hypoxic stress, the obstruction of protein folding serves as the primary cause of protein aggregation, prompting eukaryotes to develop unfolded protein responses as a regulatory mechanism (20,21). In the current study, a comprehensive review of the mechanisms involved in hypoxia-induced aggregation of unfolded and misfolded proteins, and the cellular strategies relying to this phenomenon is presented.
The number of large multidomain proteins notably increases from prokaryotes to eukaryotes. These proteins exhibit diverse conformations, and as their protein configurations become more complex, the possibility of misfolding increases (22). Hydrophobic amino acid residues, unstructured regions in folding intermediates and misfolded proteins are often exposed to solvents, leading to aggregation (23). Aggregates are primarily driven by liquid-liquid phase separation (LLPS) or hydrophobic forces, depending on the concentration (24). While most aggregates are amorphous, the aggregation of certain proteins leads to the formation of amyloid fibers characterized by β strands normal to the long fibril axis (cross-β structure) (25). Before fiber formation, amyloid often exists in an oligomeric state, and both types of aggregates play crucial roles in diseases (26). For instance, cerebral blood flow decreased in patients with early Alzheimer's disease (AD) (27). Increased binding of oligomeric β-amyloid protein (Aβ) to ROS leads to vasoconstriction around brain cells, contributing to decreased cerebral blood flow, which may initiate a cascade reaction involving amyloid Aβ itself or the fibrous Aβ, which is important for driving cognitive decline (27,28). Thus, it is necessary to understand the mechanisms underlying hypoxia-induced protein aggregation for elucidating the pathogenesis of neurodegenerative disease and developing intervention strategies.
Chaperones
Molecular chaperones play an important role in maintaining protein homeostasis, and assist other proteins in acquiring functionally active conformations without affecting their final structure. Different types of molecular chaperones receive newly synthesized protein chains from ribosomes to ensure effective folding and minimize aggregate formation by guiding them through appropriate folding pathways (26). As proteins are structurally dynamic, proteostasis occurs via a network of chaperones and protein degradation mechanisms that continuously monitor the proteome (29,30). Chaperones help prevent chain compaction and misfolding, and facilitate the removal of protein aggregates through lysosomal-autophagy degradation (31). Before degradation, the depolymerization of aggregates is cooperatively carried out by heat shock proteins (Hsps) such as Hsp70, Hsp110 and Hsp40 (32,33). The clearance pathways involving proteasomes and lysosomes are intricately linked to the Hsp70 and Hsp90 chaperone systems through specialized ubiquitin ligases such as the co-chaperone C-terminus of the Hsc70-interacting protein and the BAG domain (34,35).
However, under hypoxia conditions, the regulatory network of protein homeostasis is disrupted, and numerous molecular chaperones are affected by hypoxic stress. Nguyen et al (36) observed notable global reductions of ATP-dependent Hsp70 and Hsp90 (83 and 78%, respectively) after 24 h of hypoxia treatment. Conversely, the protein expression of the ATP-independent Hsp27 and Hsp40 in the brain, heart and muscle remained constant throughout the 24-h hypoxia treatment. However, with prolonged hypoxia, the expression of the Hsp27 and Hsp40 genes in these tissues was also reduced, suggesting that the protein expression of these chaperones may also eventually decrease under hypoxia. These results suggest that energy conservation is prioritized over cytoprotective protein chaperoning in naked mole-rat tissues during acute hypoxia. Although ATP-independent partners do not require ATP to regulate their functional cycle passive histone aggregation (37), aggregate bursts under low oxygen stress also suggest that these ATP-independent partners cannot remedy the homeostatic imbalance caused by the energy gap. In fact, the effects of hypoxic stress on protein chaperones are not machine-made, for example, C2C12 cells induce Hsp70 gene expression through a similar mechanism to heat stress during acute hypoxia (38). However, macrophages exposed to 5% oxygen for 24 h notably reduced Hsp70 expression and recovered after reoxygenation (39). Proteomics indicated that Hsp72 downregulation in the cerebral cortex of rats after 5 days of hypoxia reached its lowest level (40). In addition, the Hsp90 chaperone family TRAP1 has been found to be frequently induced in tumors and regulate energy metabolism after HIF-1 stabilization (41), and hypoxia can also reduce the transcription of cyclin B1 in liver cancer cells through Hsp90 (42). These contradictory results may be due to differences in the function and distribution of molecular chaperons, and the crosstalk between hypoxia stress and chaperons may need further exploration.
Disulfide bonds
Disulfide bonds are commonly found in protein domains located in the cytoplasmic membrane and enhance protein stability. The cleavage of disulfide bonds triggers the function of some secreted soluble proteins and cell-surface receptors (43). Oxidative protein folding refers to the restorative process through which proteins containing disulfide bonds transit from fully reduced and unfolded states to their original bioactive forms (44-46). Koritzinsky et al (47) used 35S labeling and suggested that the production of disulfide bonds was limited by hypoxic surroundings and that protein folding recovered upon oxygen restoration (48,49). This evidence suggested that oxygen depletion may seriously impede disulfide bonding leading to protein misfolding.
In brief, oxygen deprivation disrupts protein folding through multiple mechanisms, including inhibiting disulfide bond formation, inactivation of molecular chaperones and elevation of ROS levels (50,51). Prolonged accumulation of misfolded proteins may eventually result in the formation of pathological protein aggregates (Fig. 1). This shift contributes to the development of neurodegenerative diseases, such as AD, Huntington's disease (HD) and Parkinson's disease (PD) (3,52,53). Moreover, hypoxic-ischemic encephalopathy (HIE) occurs when the brain is exposed to oxygen deprivation and ischemia. Newborns often experience HIE due to birth asphyxia, causing an unfavorable prognosis owing to cerebral dysfunction, neuronal cell death and neurological deficits. Notably, marked molecular and subcellular changes observed in the brain cells of patients with HIE include protein misfolding, aggregation and organelle damage (54). The disruption of protein homeostasis is also closely related to cardiac hypertrophy, cardiomyopathy and heart failure caused by cardiovascular hypoxia (55). Soluble protein oligomers have been observed in the myocardial cells of patients with idiopathic dilated cardiomyopathy, non-ischemic cardiomyopathy, or hypertrophic heart disease (56). Similarly, aggregation of abnormal and ubiquitinated proteins has been detected in the heart of individuals with dilated cardiomyopathy or ischemic heart disease (57). Pattison et al (58) previously demonstrated that the expression of ectopic gene that containing 83 glutamine repeats in cardiomyocytes promoted the cohesive accumulation and aggregation of pre-glutamine amyloid oligomers, increasing protein deposition, cardiac muscle cell death and heart failure.
Proteostasis in condensate aggregation
Cellular proteostasis is tightly controlled by a network of molecular chaperones. In addition to counteracting abnormal folding and aggregation by directly binding to misfolded proteins (59), chaperones also assist the ubiquitin-proteasome system (UPS) (60) and the autophagy-lysosome system in degrading aggregators for proteostasis (61).
The lysosomal-mediated autophagy degradation pathway is a major hunter for clearing protein aggregates, especially in neurodegenerative diseases (62). Most neurodegenerative diseases involve pathological abnormal protein aggregates, developing neurofibrillary tangles. For example, Aβ and C-terminal fragments of the amyloid precursor protein in AD, mutant α-synuclein in PD, polyglutamine-expanded huntingtin in HD, and mutant superoxide dismutase 1 and TAR DNA-binding protein 43 (TDP-43) in ALS (63-65). These protein aggregates mainly target the autophagy lysosomal degradation pathway, and chaperone proteins play a key role in this process. Specific aggrephagy receptors have been reported in yeast S. cerevisiae (Atg19) and C. elegans (SEPA-1 and EPG-7) (66-68). Recently, Ma et al (69) reported the function of the TRiC subunit chaperonin-containing TCP-1 subunit 2 (CCT2) in aggrephagy in mammals and yeast. CCT2 promotes autophagosome incorporation and clearance of protein aggregates with little liquidity by interacting with ATG8s and aggregation-prone proteins independent of cargo ubiquitination. The dual function of CCT2, as a chaperone and an aggrephagy receptor, enables double-layer maintenance of proteostasis.
Cellular stress and aging can lead to a decrease in protein homeostasis. In addition to the inhibition of protein chaperone activity by hypoxia metabolism, notably, hypoxia-reoxygenation treatment dysregulates key molecules that maintain autophagy-lysosomal flux in primary human trophoblasts, notably reduced autophagosomes and autolysosomes (70). The expression of ubiquitin 26S-proteasome E3 ligase, autophagolysosomal degradation related mRNA transcripts and proteins, and integrated stress response markers were also decreased after 12 days of hypoxic feeding (71).
The UPS system is strongly associated with regulating biomolecular condensation (60). More specifically, ubiquitin and other post-translational modifications act as agents of phase separation, and are essential for the formation of condensates and ubiquitin-proteasome system activity (5). It is noteworthy that previous studies demonstrated that polyubiquitin chains can function as multivalent molecules that can drive either the assembly or the disassembly of condensates via interactions with various ubiquitin-binding proteins (72,73).
Unfolded protein reaction (UPR) in the regulation of unfolding/misfolded protein aggregation
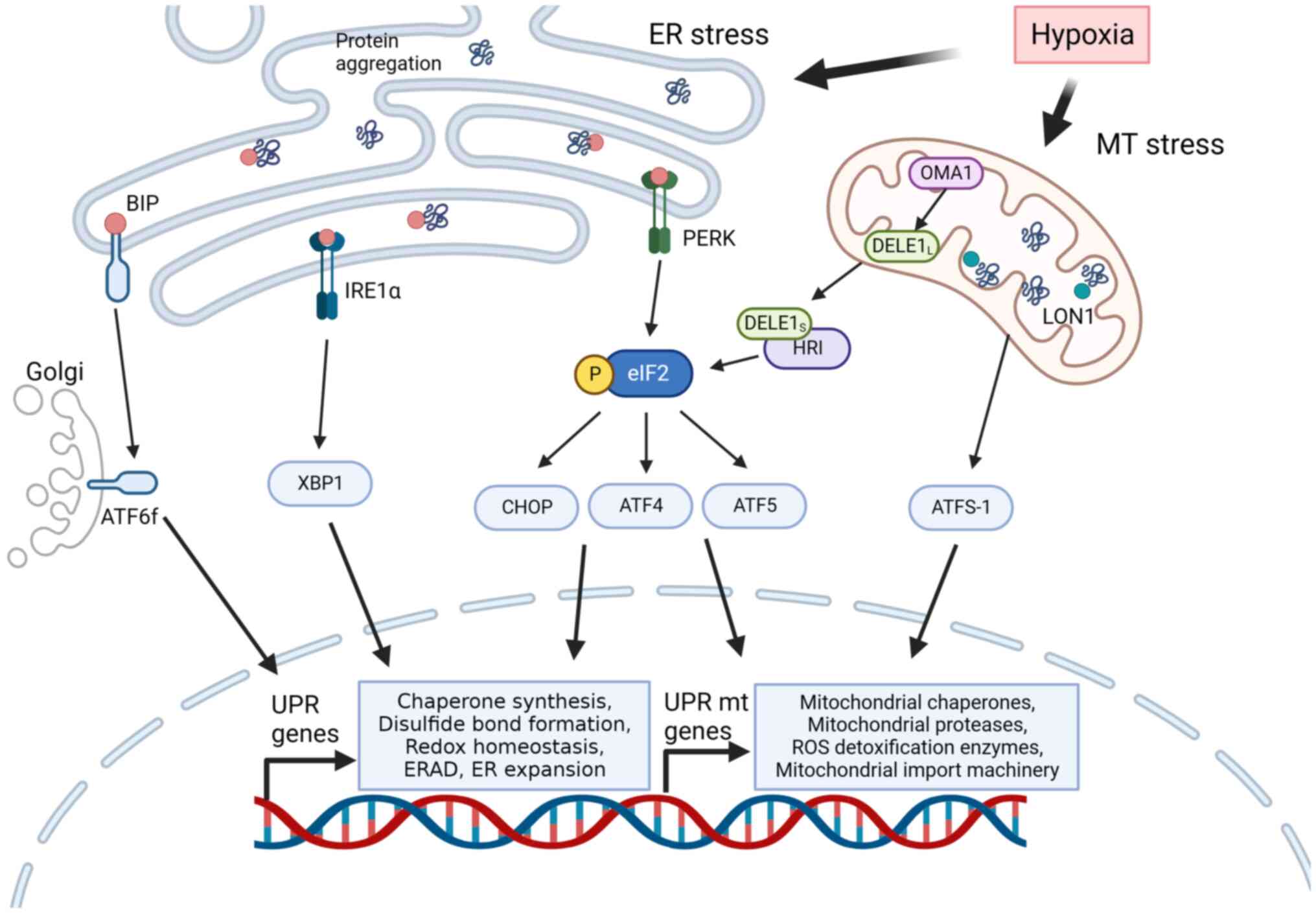
Cellular responses to hypoxia primarily aim to enhance cell survival and restore oxygen equilibrium. In the context of uncontrolled protein folding, the accumulation of unfolded or misfolded proteins within the ER or mitochondrial space leads to activation of UPR (50,53). Through its distinct signalling network, the UPR pathway restores protein homeostasis, alleviates the burden of protein aggregation and maintains cell viability (74-78). The heavy-chain-binding protein (BIP), a member of the Hsp70 family, is a crucial chaperone that triggers UPR activation. BIP enters the ER by binding to hydrophobic amino acids to prevent incorrect folding and polymerization of the polypeptide chains. This is followed by ATP binding and subsequent release of the bound polypeptides through ATP hydrolysis (79). Environmental stress leads to misfolded proteins accumulating, causing the release of BIPs (80). The released BIPs undergo phosphorylation and polymerization, triggering the activation of protein kinase R (PKR)-like ER kinases (PERKs) and inositol-requiring enzyme-1 (IRE1) (81). Additionally, activating transcription factor (ATF) 6 is switched to the Golgi apparatus and convered to soluble and active cytoplasmic ATF6 (82-84). These PERK, IRE1 and ATF6 sensors constitute three distinct signalling pathways within the UPR (80,85). Hypoxia induces BIP expression in both cancer and endothelial cells (86-88). Hypoxia can activate the PERK signalling pathway in various models (89-91), and the phosphorylation of eukaryotic initiation factor 2 (eIF2) mediated by PERK was observed within minutes of hypoxic exposure, with a reduced response rate as the oxygen concentration increased (92). To alleviate ER stress, UPR signalling inhibits protein aggregation by reducing protein synthesis flux and activating the transcriptional program of molecular chaperones.
The hypoxia-mediated UPR has been well demonstrated in the tumor microenvironment, and exposure of solid tumors to intermittent hypoxia may lead to high ROS levels and UPR activation (93-95). For example, increased ATF4 expression has been shown in numerous hypoxic and nutrient-deprived tumors (96) and can mediate autophagy under hypoxia (97). Immunohistochemical staining demonstrated increased expression of ATF4 in hypoxic, perinecrotic regions distal to the tumour vasculature, consistent with a nutrient-deprived mechanism of translational activation. In addition, the distribution of p-eIF2a and p-GCN2 signal demonstrated considerable association in serial sections, consistently, spontaneous mouse tumours also contain greater levels of p-eIF2a and ATF4 than corresponding normal tissue (98). PERK and ATF4 protect glioblastoma cells exposed to cyclic hypoxia or radiotherapy from oxidative damage (99,100). In human cervical cancer, PERK activation leads to the accumulation of oncogenic lysosomal-associated membrane protein 3, thus increasing the aggressiveness of these cells (99).
UPR in mitochondria
Mitochondria are the primary consumers of oxygen within cells. Early mitochondrial dysfunction is implicated in numerous hypoxic diseases such as cancer and neurodegenerative diseases (17,101,102). The efficiency of mitochondrial oxidative phosphorylation is markedly reduced under hypoxic conditions due to mitochondrial perinuclear localization and fragmentation mediated by CHCHD4 (103-105). Mitochondria contain their inherent genetic information and rely on stress response systems to translate and fold encoded proteins, and refold nuclear-encoded proteins (106). Maintenance of protein homeostasis in this organelle involves unique molecules such as Hsp60 and the peptidase lon peptidase 1 (106,107). Under hypoxic conditions, mitochondria can also experience unfolded or misfolded proteins aggregating. For example, using C. elegans, Kaufman et al (9) identified 65 preferentially insoluble mitochondrial proteins and 110 generally insoluble mitochondrial proteins during hypoxia, and reported that the abundance of hypoxia-induced mitochondrial protein aggregates (HIMPA) increased notably with the severity of hypoxia. Additionally, Yan et al (108) reported that disruption of mitochondrial proteostasis and mitochondrial protein aggregation are early processes involved in hypoxia in C. elegans. Like in the ER, mitochondria also activate their own UPR, which is known as the mitochondrial UPR (UPRmt). The UPRmt is classically considered as a transcriptional response that increases the expression of mitochondrial chaperones to protein misfolding and aggregation in mitochondria (109-111). In C. elegans, the UPRmt was found to be regulated by sensitizing transcription factor associated with stress 1 (ATFS-1), which is a transcription factor within mitochondrial and nuclear localization sequences, and dual subcellular localization. ATFS-1 is transported into the mitochondrial matrix and then degraded by LON proteases under steady-state conditions. The transport of ATFS-1 is downregulated in mitochondrial dysfunction, and ATFS-1 is subsequently transported to the nucleus to stimulate transcriptional responses (111,112). Additional regulatory mechanisms may exist in mammalian cells, with ATF5 acting as a functional ortholog of ATFS-1 (113). In addition, ATF4 and the C/EBP homologous protein activating are important in the activation of UPRmt (114,115). Activation of UPRmt to mitochondrial stress in cancer could maintain mitochondrial integrity and tumor growth (116). A recent study by Sutandy et al (117) showed that UPRmt signaling is prompted by the release of two individual signals in the cytosolmitochondrial ROS (mtROS) and mitochondrial protein precursors in the cytosol, leading to the release of HSF1 by Hsp70, which results in nuclear translocation and transcription of UPRmt genes (117).
The expression of these transcription factors is mediated by eIF2α kinase phosphorylation (118). Recently, Guo et al (119) delineated the relationship between mitochondrial stress and the relay of ATF4. Heme-regulated initiation factor 2 α kinase (HRI) is a necessary eIF2 kinase for this relay. A genome-wide CRISPRi screen identified two upstream signaling factors for HRI: The OMA1 zinc metallopeptidase (OMA1), as a mitochondrial stress-activated protease, and the DAP3 binding cell death enhancer 1 (DELE1) associating with the inner mitochondrial membrane. Mitochondrial stress results in DELE1 cleavage by OMA1 and its accumulation in the cytosol, which interacts with HRI and increases eIF2 kinase activity. These results indicated that the UPRmt and UPR signaling pathways can been interlinked via eIF2α (Fig. 2) (109-111).
HIMPA consistently alleviates hypoxia-induced cell death, and UPRmt activation had the same effect. However, UPRmt is not necessarily protective against hypoxia-induced cell death (108). It is the overactivation of UPRmt that can induce cell death as in the case of UPR (118), and the relationship of HIMPA with UPRmt and its crosstalk with UPR needs to be explored further.
3. Hypoxia-induced biomolecular condensates assembly
Previous studies have suggested that the cytosol is not uniform in which proteins diffuse freely, but rather formed biomolecular condensates with phase separation (52,120). Previous studies have shown that cytoplasmic proteins or RNAs are organized into distinct biomolecular condensates (52,121,122). These condensates, also known as organelles without membrane, employ the cytoskeleton for targeted transport. These proteins serve as the center for biochemical reactions, act as signaling hubs and execute a wide range of physiological functions when required (123). LLPS is a principal method for condensing of biological macromolecules. This gives rise to a resemblance of 'order' within the seemingly 'chaotic' cells and a new framework for organization of macromolecules (121,124).
Inside the cell, LLPS formation first requires that the macromolecule (protein, DNA, or RNA) in the solution reaches a certain concentration threshold, knowing that an excessive threshold can induce phase separation under suitable pH and temperature conditions (121,125). Biological macromolecules exist in two forms: A diluted state in solution and a concentrated state in 'droplets' (126), and the two forms are dynamically interchangeable as the relevant conditions shift (3,127,128). Cells can regulate the concentration at which specific proteins form droplets by altering post-translational modifications (129), and then assemble into biomolecular condensates by recruiting relevant macromolecular components. A protein or RNA that acts as the phase separation scaffold or starter in the assembly process is called the 'scaffold molecule', and the assembled material is called the 'client molecule' (130). The currently recognized 'scaffold-client' molecular model of the assembly of biomolecular condensates is described below (121,131). In addition, condensates are also controlled by the protein quality control machinery, which includes molecular chaperones and protein degradation systems (132). With enriching in specific proteins and other components, condensates can execute various biological functions in different cellular compartments. These effects can be attributed to condensation including the promotion (133) or inhibition of biochemical reactions (134), reduction of protein concentrations (135), detection of fluctuating in the environment (136) and mechanical forces (137).
In hypoxic environments or hypoxia-related disease models, certain biomolecular condensates are equipped with cellular regulatory functions and are used to regulate the metabolism of cells or maintain their survival (Table I). This section summarizes the activation mechanisms and physiological functions associated with these hypoxia-induced condensates.
SGs
SGs are assemblies of non-translating messenger ribonucleoprotein granules, various non-membrane-bound cellular compartments that contain high concentrations of proteins and RNA (138), and are close to UPR (139). The formation of SGs is facilitated by interactions between mRNAs and mRNA-binding proteins, translation initiation factors, the 40S ribosomal subunit (a myriad of RNA-binding proteins) and translationally stalled mRNAs (139,140). Once the cells return to a normal and non-stressful environment, SGs disperse and protein translation is reinstated (141). Eukaryotic cells use SGs to redirect limited resources from protein synthesis to survival and stress resistance.
The core of the SG central node of this network incorporates the G3BP SG assembly factor 1 (G3BP1), which serves as a molecular switch instigating RNA-dependent LLPS in response to elevated concentrations of free RNA in cells. G3BP1 is also capable of modulating LLPS propensity via three different inherently disordered regions. The core SG network can be simultaneously reinforced or weakened by altering G3BP1-binding factors (142). The assembly activation cues of SGs coalesce with UPR signals to create networks that maintain protein homeostasis (139,143). The conventional assembly process of SGs is mediated by eIF2 phosphorylation. The eIF2 kinase family includes PERK, PKR, general control non-depressible 2 (GCN2), and HRI (144,145). In a hypoxic environment, eIF2 phosphorylation is induced by PERK and activated through UPR signaling or the OMA1-DELE1-HRI pathway, which is initiated by UPRmt (119). The phosphorylation state of eIF2 is regulated by its interaction with eIF2β, and this interaction inhibits the conversion of GDP to GTP by eIF2β, resulting in a decrease in the concentration of ternary complex eIF2-GTP-tRNA Met (146,147). Consequently, the RNA-binding protein TIA1 and T-cell-restricted intracellular antigen-associated protein (TIAR) stimulate the formation of the noncanonical 48S preinitiation complex (148). This complex, unable to recruit the 60S ribosomal subunit translation, can be used for SG assembly (148-150).
In addition to being mediated by UPR activation, hypoxia causes the assembly of SGs through several other pathways. Hypoxia is frequently associated with nutrient scarcity, and mammalian cells can sense alterations in amino acid levels through the GCN2 and mTORC1 pathways (151). Amino acid deprivation inhibits mTORC1-mediated protein translation but stimulates angiogenesis via the GCN2-ATF4 amino acid starvation response pathway, that is independent of HIF-1 (152,153). GCN2 also promotes eIF2 stimulation and collaborates with PERK to shield hypoxic cells from apoptosis (154). Furthermore, hypoxia generally triggers type I interferon (IFN) pathway inhibition and reduces IFN secretion, which could lead to uncontrolled double-stranded RNA (dsRNA) expression (155). As a stressor, dsRNA can incite the phosphorylation of eIF2α via PKR. This phosphorylation results in the formation of SGs, which serve as an antiviral core (156). To summarize, hypoxia instigates the activation of SG assemblies (Fig. 3).
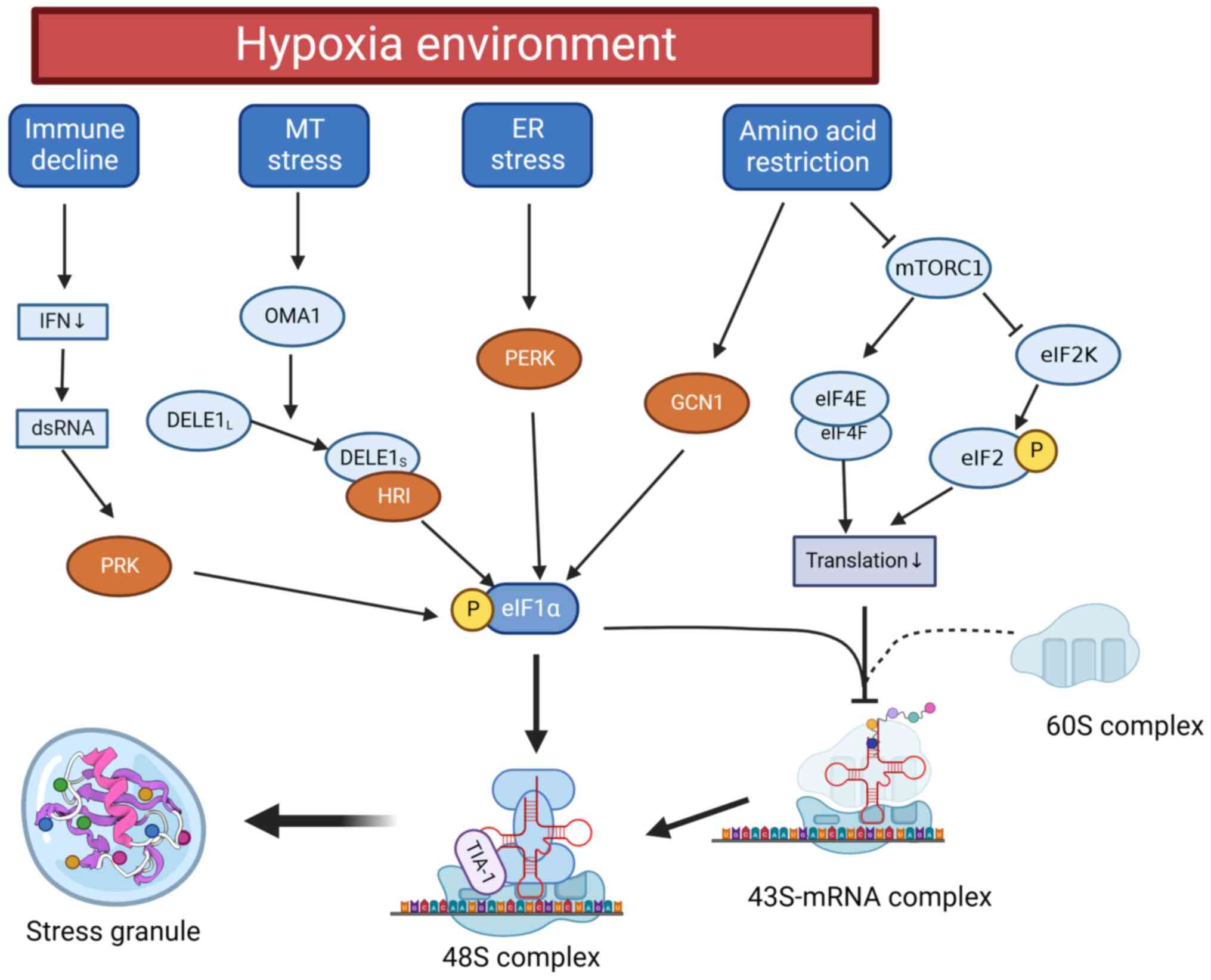
Hypoxia-induced SG assembly effectively improves cell viability, which has been well demonstrated in the hypoxic microenvironment of cancer. Apoptosis-related molecules that accumulate within SGs assembled by cancer cells manifested antiapoptotic effects (157), and the development of hypoxia-induced SGs causes drug resistance in cancer (158). By pharmaceutically impeding hypoxia-induced SG formation in HeLa cells, Timalsina et al (159) managed to decrease drug resistance in hypoxic microenvironments. A study by Attwood et al (160) showed that hypoxia increased the number of late apoptotic/necrotic glioblastoma cells during the raloxifene-induced delay in SG dissolution. Liu et al (161) provided that hypoxic conditions could result in FUS-circTBC1D14-associated SG formation in the cytoplasm after PRMT1 modification, thus contributing to the maintenance of cellular homeostasis and promoting tumor progression in triple-negative breast cancer.
In rodent models, SGs were found to protect hepatocytes against hypoxia-induced damage by reducing apoptosis. With the increased expression of the SG marker proteins G3BP1 and TIA-1, the degree of liver injury, HIF-1α and apoptosis induced by acute liver failure decreases (162). In addition, Hu et al (163) found that impaired SGs are important in the pathogenesis of spinal muscular atrophy.
It is noteworthy that nematodes and rat cardiomyocytes produced characteristic SGs in mitochondria stimulated by sublethal hypoxia. Mitochondrial SGs are involved in early mitochondrial pathology and are closely associated with UPRmt (14).
P-bodies
P-bodies are also a type of biomolecules participating in phase separation (164). The structure of P-body is similar to that of SGs, and P-body shuttle RNA binding proteins and mRNAs between the two condensates (165,166). Usually, SGs uniquely house certain translation initiation factors, while P-body specifically abound to factors associated with mRNA degradation and decay, leading to functional differences (164,167). In the presence of hypoxic stress, SGs can maintain cell survival (168), while P-bodies seemed to be more inclined to regulate hypoxia-related signaling molecules.
Past research has demonstrated that hypoxia can induce RNP granule formation in C. elegans oocytes, and RNP foci are similar to the RNA-related functions of P-bodies (169). Saito et al (16) reported that HIF-1α was upregulated by the microRNA (miR)-130 family during hypoxia. The miR-130 family was increased under hypoxia, and their target was DDX6 mRNA, a component of the P-bodies. These results reveal a new translational mechanism of HIF-1α and P-bodies in hypoxic stress (16). The USP52 protein and HIF1A mRNA were found to colocalize with cytoplasmic P-bodies, suggesting that P-bodies recruit HIF1A mRNA for assembly through LLPS. The P-body component USP52/PAN2 can enhance the stability of HIF-1α mRNA, which is crucial under hypoxic conditions (170). Moreover, HIF1A mRNA localizes to P-bodies following microtubule disruption for a short period of translational repression (171). These findings suggest that P-bodies contribute to the regulation of HIF1A mRNA stabilization and protein translation, which are critical for hypoxic signaling and cellular hypoxic response (17).
Glycolytic body
Metabolic flux is an important intracellular change that occurs during hypoxic stress. When cellular oxidative phosphorylation is impaired by hypoxia, glycolysis becomes the primary source of energy (172,173). Although the glycolytic pathway has a shorter energy supply pathway, the total amount of ATP produced is lower than that produced during oxidative phosphorylation (174). To meet the ATP required for survival and speed up the flow of glycolysis, cells integrate the enzymes required for glycolysis and other scaffold proteins through LLPS to form a special biomolecular condensate, called a glycolytic body (G-body) (15,175).
Under hypoxic conditions, glycolytic enzymes are compartmentalized into cytoplasmic structures (176), and analogous condensates form were also found in C. elegans neurons (177). Therefore, Jin et al (15) demonstrated that under hypoxic conditions, cells assemble non-membrane organelles that include glycolytic enzymes, called G-bodies. They also found that glucose consumption increased, and that the level of glycolytic intermediates decreased in cells with G bodies. It is noteworthy that the formation of G-bodies increases the glycolytic output in hypoxia (15) via glycolytic enzymes such as phosphofructokinase, pyruvate kinase, acetyl-CoA carboxylase and yeast pyruvate kinase Cdc19 (178-181). These enzymes can catalyze the rate-limiting step in glycolysis and be utilized to increase the glycolysis rate under hypoxic conditions. While the mechanism of G-body activation has not been elucidated. Gregory et al (182) detected hundreds of RNA-binding proteins in G-bodies using genomic and proteomic methods. The failure of nonspecific endonucleases to maintain the structural integrity of G-bodies suggests that the assembly of G-bodies replying to hypoxia is likely mediated by an RNA-dependent phase separation mechanism (182). The enzymes involved in the formation of G-body aggregates follow a specific order post-nucleation, and the entry of each metabolic enzyme into the G-body is tightly regulated (183). The multiple glycolysis enzymes within phase separation may function to enhance the activity and increase the reaction rate in energy production, thereby forming 'metabolons' during hypoxic stress (184).
Notably, cells that are unable to form G-bodies undergo abnormal division and yield nonviable daughter cells during hypoxia, and the formation of G-bodies represents a conserved adaptive response that maintains the energy requirements of the cells (15).
Lipid droplets (LDs)
Fatty acids consist a major fuel in various cells. The depletion of oxygen substrate severely inhibits the fatty acid β oxidative energy pathway of the cell, and the accumulated excess fatty acids are transformed into triglycerides for storage (185,186). The ER participates in synthesizing these triglycerides, which are subsequently stored in biomolecular condensate called LDs (187). LDs are dynamic lipid compartments that can effectively manage fluctuating cellular lipids. Following oxygen restoration and activation of fatty acid oxidation, LDs are broken down by neutral lipase, and the liberated fatty acids serve as substrates for mitochondrial oxidation, leading to energy production (188). LDs contain core lipid components, and are surrounded by an amphipathic lipid layer (189). Almost all organisms synthesize LDs, whose formation is initiated by the synthesis of neutral lipids (NLs) (190). Overnutrition or various stressors prompts cells to produce NLs in the ER bilayer (191,192), where the synthesized NLs mix with phospholipids on the membrane and diffuse in the ER bilayer (193). When the NL concentration exceeds the nucleation threshold, LLPS drives LD formation to prevent NL accumulation in the ER membrane (194).
Hypoxia-induced LDs were initially observed in cancer cells (195). They may require substantial lipids for biosynthesis, and lipid-derived bioactive molecules for cytomembrane formation and a high level of cell proliferation (196,197). It has been demonstrated that lipid formation via HIF-1α mediates reductive glutamine metabolism instead of pyruvate-mediated acetyl-CoA production in cancer cells (198). Thus, a high LD content was closely related to transcription driven by hypoxia in hypoxic cancer cells. LDs are associated with various malignant phenotypes (198). Mounting evidence supports the diverse roles of LDs in cancer cells responses to stress conditions, such as maintaining ER homeostasis (199), clearing ROS (200) and preventing drug resistance (201), all of which are crucial in the maintenance of homeostasis in cancer cells.
LD formation and degradation are controlled by numerous enzymes and LD-associated proteins. Hypoxia-inducible LD-associated protein (HILPDA) is a paramount LD-associated protein induced by HIF-1 and fatty acid expression. It localizes in the LDs of several cell types, and is situated near the ER and LDs within cells. HILPDA directly inhibits the activity of adipose triglyceride lipase via physical interaction and encourages LD accumulation by stimulating triglyceride synthesis (202,203). These findings suggest that under hypoxic stress, not only proteins and RNA, but also lipids can be orchestrated to assemble into specific molecular biopolymers for survival.
Other protein condensates associated with hypoxia adaptation
Hypoxic stress can induce the formation of protein condensates, which play a role in promoting basic biochemical processes. For instance, prolyl hydroxylases are involved in regulating molecular responses to oxygen availability. These proteins hydroxylate HIF-α, enabling its ubiquitination and degradation (204,205). Increased expression of HIF can lead to the generation of ROS, which modulates HIF-α stabilization in conjunction with prolyl hydroxylase domain proteins (PHD) (206,207). The PDH family has a function in regulating HIF through the condensation of PDH3, a protein expressed in response to oxygen deprivation that contributes to neural cell death. PDH3 forms subcellular condensates in the presence of oxygen, but its condensation is notably decreased under hypoxia (208,209). The formation of PDH3 condensates relies on microtubules and involves the integration of components from the 26S proteasome, chaperones and ubiquitin. The PHD2 condensates exhibit liquid characteristics similar to other condensates (210). When PHD3 is actively expressed under normoxia, it leads to the condensation of proteasome components, triggering apoptosis in HeLa cells. Apoptosis occurs in cells prone to PHD3 condensation and is observed before apoptosis (210).
Recently, Theodoridis et al (211) discovered that hypoxia-induced cell acidification could induce the aggregation of certain amyloid proteins in the nucleus. These proteins are not unfolded proteins but rather formed through phase separation of a class of long-chain non-coding RNAs derived from a specific site of stimulation within the ribosomal gene spacer (211). Local nuclear translation under stress conditions is crucial under various physiopathological conditions. Amyloid bodies enhance local nuclear translation during stress, suggesting that aggregates, similar to liquid condensates, can facilitate complex biochemical reactions (211,212). (However, a further detailed assessment is required to determine the degree to which soil-like condensate formation occurs under stress.
In conclusion, cells initiate the assembly of biomolecular condensates to sustain cell survival and regulate metabolism in response to hypoxic conditions. A concise overview of the crucial components and biological functions of hypoxic-related biomolecular condensates is presented in Table I, which can provide valuable information for future research on hypoxic-related diseases.
4. ATP drives protein dissolution and biomolecular condensation assembly
Exposure of cells to hypoxia leads to the impairment of cytochrome C oxidase activity, resulting in the generation of ROS and the inhibition of ATP synthesis (17,219,220).
ATP-driven protein chaperones and molecular motors play crucial roles in activating molecular condensation and regulating solubilization. ATP-dependent depolymerases are responsible for dissolving aggregates and reordering them for refolding or degradation (221,222). In yeast, the ATP-generating enzyme Cdc19 is incorporated into SGs to form reversible amyloid structures under stressful conditions (223,224). Rapid re-solubilization of these amyloids is essential for ATP generation and subsequent breakdown of SGs (180). Increasing energy metabolism enhances Cdc19 re-solubilization in yeast, while the recruitment and aggregation of the ATP-dependent chaperones Hsp104 and Ssa2 can enhance the efficiency of solubilization (225).
The formation of misfolded protein aggregates is regulated by molecular chaperones. Small Hsp sequesters such as yeast Hsp26 can promote misfolded protein aggregating, facilitating subsequent refolding (226). In yeast, the Hsp70 protein cooperates with Hsp104 disaggregate to solubilize aggregated proteins with ATP (227). Energy-dependent processes or molecular machinery also participate in regulating the extent of fiber formation within condensates. These processes could restrict the formation of structures when dynamic condensates are required, and facilitate their formation and growth when static condensates are necessary. This explains the reason numerous higher-order assemblies contain molecular chaperones, ATP-dependent depolymerases and molecular motors (131,228). A previous study in newborn rats subjected to unilateral carotid ligation and then exposed to hypoxia for 80 min showed varying levels of hsp72 mRNA expression in the area of ATP reduction induced during hypoxia recovery (229). In renal epithelial cells, Hsp72 expression is increased in response to ATP depletion, especially after thermal preconditioning (230). Other studies have shown that hypoxia/reoxygenation or ATP depletion can reduce Hsp60 levels, induce Bax transfer to mitochondria and cause apoptosis (231). Although it is unclear whether ATP produced from glycolysis under hypoxia is inadequate to support molecular chaperones, these results also suggest a strong link between hypoxia-induced ATP depletion and changes in protein chaperones.
With the role of ATP in driving enzymatic activity, more direct evidence arises from the hydrophilic tripolyphosphate and a relatively hydrophobic adenosine ring, which provide ATP with amphiphilic properties (232,233). Patel et al (234) demonstrated that ATP could prevent the liquid-liquid phase separation of FUS, and even dissolve previous droplets within the liquid phase compartment. This effect was also observed for TATA-Box Binding Protein Associated Factor 15, heterogeneous nuclear Ribonucleoprotein A (hnRNPA) 3 and phosphogluconolactonase 3 in the liquid phase compartment. Increasing the ATP concentration to 2 mM in the chamber achieved a similar solubilization effect by inhibiting protein aggregate formation and maintaining protein solubility (234). These findings provide a new direction for understanding disorders associated with aberrant amyloid aggregation or a hypoxic environment.
5. Transformation of aggregates and condensates
Previous studies have often focused on either the assembly of aggregates or the formation of healthy molecular condensates (13-15,53-55). However, they have rarely considered them together, resulting in conceptual separation between these macromolecular structures. Protein aggregates and condensates are closely related because they both involve higher-order assemblies with stoichiometric ratios (3).
Under different conditions, protein aggregates or condensates can originate from intermediate clusters as aggregation or droplet precursors. Another possible mechanism of protein aggregation involves the initial formation of a droplet as an intermediate aggregate, which undergoes a transition into a solid state. Recent studies focusing on proteins such as FUS, hnRNPA1, TDP43 and Tau associated with neurodegenerative diseases including ALS, AD and PD have shown that liquid-phase condensation precedes protein aggregation and amyloid formation (235-237). However, multiple studies have suggested that cross-β (or amyloid) interactions are involved in the formation of protein aggregates, and amyloid fibril formation is frequently found in phase-separated proteins in vitro (238,239). This means that the condensates can be mutually converted to some extent.
The assembled molecular condensate itself can also be transformed into a more solid-like state, a process known as aging or hardening (136,240,241). The aging pathway of agglomerates involves the gradual transition of glass-like condensates from a fluid state to a more solid-like state. These glass-like condensates undergo continuous changes in their properties but do not fully solidify (242). Their behavior is influenced by multiple factors, such as temperature and density, which affect their propensity for undergoing transitions (243). Over time, glass-like condensates show reduced elasticity and shrinkage, indicating an increase in molecular contacts and aggregation (244,245). Another method of transformation is gelation, with weak or strong interaction forces, and coacervate components result in the formation of a physical gel such as the gel formed by the extracellular matrix protein elastin (246,247). High concentrations of proteins, lack of physiological chaperones and low water content are factors contributing to condensate aging (248). Conversely, it has been revealed that cells can prevent condensate aging by altering the condensate composition (249), thereby minimizing the potential for conformational changes in the protein aggregation pathway. This regulatory process is often associated with energy-consuming processes (249). However, the regulatory mechanism that prevents aggregate aging is impaired in a hypoxic environment with a notable decrease in ATP levels (37,90,221,228). Additionally, the cosolvent effect of ATP is weakened under these conditions, resulting in an increased propensity of proteins to aggregate. Several factors collectively contribute to the aging of condensates (Fig. 4).

In conclusion, under physiological conditions, dynamic equilibrium is maintained between the liquid and solid phases within cells through the vigilant regulation of an intricate network of molecular chaperones and regulatory mechanisms. However, in various disease associated with hypoxia or hypoxic stimulation, the function of molecular chaperones is disrupted, leading to the accumulation of misfolded proteins and subsequent formation of numerous aggregates, thereby compromising protein homeostasis. It is hypothesized that this phenomenon is closely linked to hypoxia-induced ATP depletion.
6. Therapeutics targeting biomolecular condensation and protein aggregation
Therapeutic strategies aimed at preventing aberrant protein aggregation and aging of biomolecular condensates have shown promising results in managing ailments, particularly neurodegenerative diseases (250). Currently, the US FDA has endorsed a broad array of drugs capable of diminishing the production of Aβ aggregates, which have been shown to be effective at prolonging patient survival (251). The treatment mechanisms of these drugs fall into the following three categories: i) Create drug-protein chaperones that mimic the activity of natural chaperones, or the synthesis of small molecules that assist in stabilizing the folded protein conformation, thereby preventing protein aggregation, and examples of such drugs include aducanumab (252) and ALZT-OP1 (253); ii) indirect disruption of the signaling pathway that governs aggregation, and several inhibitors, including CNP520 (254) and JNJ-54861911 (255), have been created to target β-site amyloid precursor protein cleaving enzyme signaling in an AD model; and iv) burgeoning approaches include regulating hypoxia signals, addressing the hypoxic state, or mitigating the chronic impact of hypoxia. Numerous small molecules are being explored for their ability to alleviate the toxic effects of protein aggregates induced by hypoxic stress. One of these molecules, melatonin, effectively prevents chemical injury and impedes the synthesis and formation of Aβ (256). The administration of vitamin B6/B12/folate and choline notably improved in hypoxia-induced memory impairment by effectively curtailing tau hyperphosphorylation at several sites associated with AD (257).
Furthermore, Li et al (258) demonstrated that mild hypoxia exposure can increase the tolerance of the brain to severe hypoxic conditions, which is termed preadaptation. This preconditioning effect also reduces Aβ levels and aids in its degradation in the brain. In a clinical context, compared with regular myocytes, preconditioning has been shown to be effective at preventing hypoxia-induced CVD by enhancing the resilience of preconditioned cardiomyocytes against hypoxic injury (258).
Within the context of cancer models, a study revealed that LLPS which alters some of the target proteins could be used as a direction for cancer treatment. Our previous study presented evidence that baicalin can serve as a potential therapy for non-small cell lung cancer by altering the solid state of cyclic GMP-AMP synthase (CGAS) in hypoxic microenvironments and thereby improving mobility (259). Additionally, hypoxia has been verified to inhibit the activation of the CGAS-stimulator of the IFN gene signaling pathway (260). P53 is known as a tumor suppressor protein. Once p53 is mutated, it will result in phase separation phase transition (261), so it provides a promising strategy to investigate new therapeutic targets focusing on p53 aggregates (262).
However, the limitations, cost and side effects of current aggregate targeted therapy remain an issue in clinical practice. It is widely acknowledged that both neurodegenerative diseases and cancer are multifactorial conditions with numerous hypotheses. Consequently, therapies targeting a single potential factor are deemed unsatisfactory (263). For instance, aducanumab, an aggregate-targeting drug for AD, exhibited adverse symptoms in ~25% of patients with amyloid-related imaging abnormalities during a comprehensive safety evaluation of a Phase 3 study involving 3,285 participants (264). The mandatory exclusion criterion for aducanumab treatment is the presence of abnormal amyloid proteins in the brain. However, available data indicate that 20-40% of patients with early-stage AD do not exhibit abnormal amyloid deposition, rendering aducanumab ineffective for these individuals (265). Furthermore, there are substantial risks associated with ALZT-OP1 due to previous clinical failures and an incomplete understanding of the pathophysiological role of Aβ in AD (253).
Therefore, in the case of hypoxic-related pathology or hypoxic stress, it is crucial to acquire a comprehensive understanding of the intricate interplay between hypoxic stress and macromolecular aggregate and condense behaviors. Consequently, an effective dual-pronged treatment strategy should be implemented: Prevention of hypoxic injury and precise intervention targeting aggregation and its behavior. This approach holds promising therapeutic prospects for clinical intervention.
7. Conclusions and perspective
Hypoxic environments are stress conditions that can lead to ATP depletion, cell acidification, disulfide bond inhibition, ER mitochondrial stress and other reactions. The accumulation of misfolded proteins induced by hypoxia promotes the development of pathological aggregates, resulting in neuronal damage. Disruption of protein homeostasis and accumulation of Aβ are directly involved in this process. Various hypoxia-related diseases, including AD, ALS, HIE, heart failure and cancer, are characterized by disturbances in protein homeostasis. Simultaneously, hypoxic pressure triggers the assembly of specific biomolecular condensates in cells. These condensates, with their distinct folding patterns, core types and recruited molecules, are responsible for specific activities related to cell viability, metabolic processes and protein homeostasis. Our understanding of these aggregates may provide deeper insights into the interplay between biochemical processes during hypoxic stress and macromolecular phase separation. Interconversion between aggregation and condensation occurs through intermediate states under specific conditions. Misfolded proteins caused by hypoxia tend to aggregate, accelerating the aging process of certain phase separation droplets.
Efforts have been made to develop small molecules that specifically target hypoxic stress and protein aggregation mechanisms. They have already been employed in clinical interventions for the treatment of hypoxic injuries and neurodegenerative disorders. A comprehensive understanding of aggregates and condensates provides insight into the biochemical processes of hypoxic stress based on LLPS, which enhances the understanding of the mechanisms underlying protein disturbances and hypoxia-related diseases. In summary, the present study may also open up new possibilities for the advancement of therapeutic strategies and drug development.
However, studies of aggregates and LLPS condensates still face limitations in clinical treatment and in vivo investigations due to the lack of suitable testing methodologies. The biological relevance of the aggregates was validated without affecting LLPS-related parameters such as protein structure and cellular physiology including pH, ionic strength and others.
Availability of data and materials
Not applicable.
Authors' contributions
WX, LF and CL conceived and designed the review. CL drafted the manuscript. BH revised and polished the manuscript. HY and KW collated the literature. Data authentication is not applicable. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
The present review was sponsored by the National Natural Science Foundation of China (grant no. 32371185), the Shanghai Science and Technology Plan Project (grant no. 23010504200), the Shanghai Talent Development Fund (grant no. 2020125), the Key Lab of Exercise and Health Sciences of the Ministry of Education (Shanghai University of Sports; grant no. 2022KF001) and the Shanghai Key Lab of Human Performance (Shanghai University of Sports; grant no. 11DZ2261100).