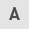Effect of metformin on the proliferation, migration, and MMP-2 and -9 expression of human umbilical vein endothelial cells
- Authors:
- Published online on: January 12, 2012 https://doi.org/10.3892/mmr.2012.753
- Pages: 1068-1074
Abstract
Introduction
Several epidemiological studies have found that diabetic patients using metformin have a lower risk of cancer in comparison to those using other anti-diabetic drugs. A case-control study by Li et al (1) reported that the risk of pancreatic cancer was 62% lower in diabetic patients who had been treated with metformin than those who had never received the drug. Other observational cohort studies demonstrated a decrease of 25–37% in cancer cases in diabetic patients treated with metformin (2,3). A study by Zhou et al (4) suggested that most of the beneficial effects of metformin are mediated through its ability to activate the AMP-activated protein kinase (AMPK). AMPK is a key sensor of the cellular AMP/ATP ratio. AMPK is activated by an increase in this proportion as a consequence of the partial inhibition of the mitochondrial respiratory chain by metformin (5). Various biological effects have been attributed to the activation of AMPK by metformin. It interferes with the action of the mammalian target of rapamycin (mTOR) that functions as part of the cellular signaling processes regulating cell growth, cell proliferation, cell motility, transcription and protein synthesis (6,7). Furthermore, the upstream regulator of AMPK is a protein kinase identified as LKB1 (8,9) which is a well-known tumor suppressor. It has been suggested that LKB1 is a critical barrier to pulmonary tumorigenesis, differentiation and metastasis (10). This fact further highlights the possible role of AMPK activation in the anticancer effects of metformin. Angiogenesis, an essential component of tumor progression, is primarily achieved through the proliferation, survival, and migration of endothelial cells (11). Angiogenesis is believed to begin with matrix metalloproteinase (MMP)-mediated degradation of the blood vessel basement membrane which contains various extracellular matrix (ECM) proteins. Subsequently, it is followed by sequential changes in vascular endothelial cells (12). MMP-2 and -9, predominately expressed in the endothelial cells, are directly involved in endothelial cell migration and vascular remodeling during angiogenesis (13,14).
Tan et al (15) reported that metformin decreases angiogenesis in women suffering from polycystic ovary syndrome (PCOS) by increasing the anti-angiogenic thrombospondin-1. In addition, metformin in a murine sponge model was found to inhibit angiogenesis by decreasing vascularization, macrophage recruitment, collagen deposition and levels of the transforming growth factor β1 (16). It can be proposed that metformin controls and reduces the progression of cancer through its anti-angiogenic effects. The effect of metformin on human umbilical vein endothelial cells (HUVECs), an established model for angiogenesis study, has not been elucidated to date. The present study seeks to address whether metformin interferes with endothelial cell functions in terms of proliferation, migration and MMP expression. In addition, we also speculated whether these effects are mediated by AMPK.
Materials and methods
Materials
HUVECs were purchased from the National Cell Bank, Pasteur Institute of Iran. Metformin was provided by the Osveh Pharmaceutical Laboratory (Tehran, Iran). Fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), TRIzol, and trypsin/EDTA 0.25% were obtained from Invitrogen (USA). Compound C, DMSO and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] were obtained from Sigma-Aldrich (USA). The Quantitect reverse transcription kit and Quantifast probe PCR+Rox vial kit were obtained from Qiagen (USA). The LDH cytotoxicity assay kit was purchased from Roche (Germany). All the other reagents used in the experiments were of analytical grade.
Cell culture
HUVECs were cultured in DMEM medium supplemented with 10% FBS. The culture was carried out at 37˚C in 5% CO2. After the cells reached a confluence of 80%, they were detached using 0.25% trypsin-EDTA. Subsequently, the cells were subcultured once again.
Endothelial cell cytotoxicity assay
The experimental procedure was conducted according to the method of Linford and Dorsa (17) for measuring the cytotoxicity and cell lysis by detecting lactate dehydrogenase (LDH) activity released from the damaged cells. HUVECs were cultured in a 96-well culture plate at a density of 1×104 cells/well in DMEM medium. After 24 h, metformin at different concentrations, compound C (10 μM), and compound C plus 3 mM metformin were added to the wells and the cells were incubated for an additional 72 h. The plates were centrifuged at 200 × g. Then, 100 μl of the LDH assay mixture was added, and the plates were incubated at 37˚C for 30 min. The LDH release was estimated at 490 nm, using ELISA (Behring ELISA Processor) and expressed as a percentage of the control. All of the experiments were performed in triplicate.
Endothelial cell proliferation assay
This assay aimed to determine whether metformin affects cell proliferation. HUVECs were seeded at a density of 1×104 cells/well in a 12-well culture plate and allowed to attach for 24 h. Next, the cells were washed twice with PBS and treated with different concentrations of metformin. HUVECs were treated with 10 μM compound C for 30 min alone or before adding metformin at the 3 mM concentration [in the present series of experiments DMSO (0.8%) was used as a vehicle]. After a 72-h incubation, the cells were washed with PBS and harvested using trypsin-EDTA. The cell count and viability were determined by trypan blue dye exclusion assay. All the experiments were performed in triplicate.
MTT proliferation assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) proliferation assay is an index of cell viability and proliferation. The cells were cultured in a 96-well culture plate at a density of 3×103 cells/well for 24 h. After being treated with different concentrations of metformin, compound C (10 μM), and compound C plus 3 mM of metformin for a further 72 h, the cells were washed twice with PBS and subjected to the MTT assay. The cells were incubated with the MTT solution at a final concentration of 0.5 mg/ml for 3 h. Subsequently, the cells were lysed in DMSO. The optical density was measured at 540 nm using an ELISA reader (Behring ELISA Processor). All of the samples were assayed in triplicate, and the mean value for each experiment was calculated. The obtained results are expressed as a percentage of the control, which is considered to be 100%.
Endothelial cell migration assay
HUVECs were cultured in a 6-well culture plate. A wound was made in the cell area using a sterile yellow tip when the cells achieved 80–90% confluence. The variation in the wound width within the experiments was approximately 5%. First, the cells were washed with PBS. Then, the cells were treated with a medium containing different concentrations of metformin and 2% FBS (2% FBS allows cell survival but not cell proliferation). After a 72-h incubation, the cells were washed twice with PBS, fixed with methanol and stained with Giemsa. The cell migration into the scratched area was photographed at a magnification of ×40. Subsequently, the cell migration was quantified by calculating the difference in the denuded area using a computerized planimetry package (Landcalc, UK). The obtained data were expressed as a percentage of the migration in the untreated endothelial cells.
RNA isolation and real-time quantitative PCR
The total cellular RNA was extracted from the cultured cells (~1×105) using TRIzol. The cells were lysed in 1 ml TRIzol and incubated at room temperature for 5 min. Then, 200 ml chloroform was added to the lysate, incubated for 3 min, and centrifuged for 15 min at 12,000 × g at 4˚C. The aqueous layer was removed, mixed with an equal volume of isopropanol and incubated for 1 h at 4˚C. The purified RNA was precipitated by centrifugation at 12,000 × g for 15 min and was finally dissolved in 50 μl diethylpyrocarbonate (DEPC)-treated water. One microgram of the total-RNA was converted to cDNA using the Quantitect reverse transcription kit (Qiagen). Real-time PCR was performed by the Quantifast Probe PCR+Rox vial kit (Qiagen) using the ABI Step One Plus Detection system (Applied Biosystems, USA). The cycling conditions were 45 cycles in two steps. An initial denaturation step at 95˚C for 3 min was followed by denaturation at 95˚C for 3 sec, and annealing-extension at 60˚C for 30 sec. For quantification, the target gene was normalized to the internal standard gene 18S. The primers were designed for detection of MMP-2 and -9 gene expression: MMP-2, forward, 5′-TTGATGGCATCGCTCAGATC-3′ and reverse, 5′-TTGTCACGTGGCGTCACAGT-3′; MMP-9, forward, 5′-GACGCAGACATCGTCATCCA-3′ and reverse, 5′-CACAACTCGTCATCGTCGAAA-3′; 18S rRNA, forward, 5′-CGGCTACCACATCCAAGGAA-3′ and reverse, 5′-GCT GGAATTACCGCGGCT-3′.
Statistics
Data are presented as the mean ± SD. One-way ANOVA was used to make comparisons between groups. A Student-Newman-Keuls post test was performed to compare the mean values between the treatment groups and the control in case the ANOVA analysis indicated significant differences. Differences between the groups were considered significant at P<0.05.
Results
Effects of metformin on LDH release from endothelial cells
HUVECs were incubated with different concentrations of the drugs for 72 h to determine whether metformin, compound C, and DMSO (as a vehicle) are cytotoxic against endothelial cells. Subsequently, the lactate dehydrogenase (LDH) release was measured. The LDH activity of the control and treated groups is shown in Table I. The LDH values among the groups were almost identical with no significant differences.
Table IEffects of metformin, compound C, and DMSO on lactate dehydrogenase (LDH) activity in HUVECs. |
Effects of metformin on endothelial cell proliferation
Incubation of the unstimulated human umbilical vein endothelial cells with different concentrations of metformin (0.5–3.0 mM) for 72 h induced a marked (P<0.05; P<0.001) and dose-dependent reduction in the number of cells (Fig. 1). Compound C was used as a pharmacological inhibitor of AMPK for evaluating the role of the AMPK pathway in the metformin anti-proliferation effects of HUVECs. Compound C, at a concentration of 10 μM, caused a 16% reduction (P<0.001) in the cell proliferation by itself. Metformin at the concentration of 3 mM produced a strong (P<0.001) inhibition of HUVEC proliferation both in the presence (37% inhibition) and absence (55% inhibition) of compound C in comparison to the related controls. However, the anti-proliferation effect of metformin (3 mM) was significantly (18%; P<0.01), but not completely reversed by compound C (Fig. 1). This inhibition did not result from a cytotoxic effect, as assessed by the LDH release from the control and the treated groups (Table I).
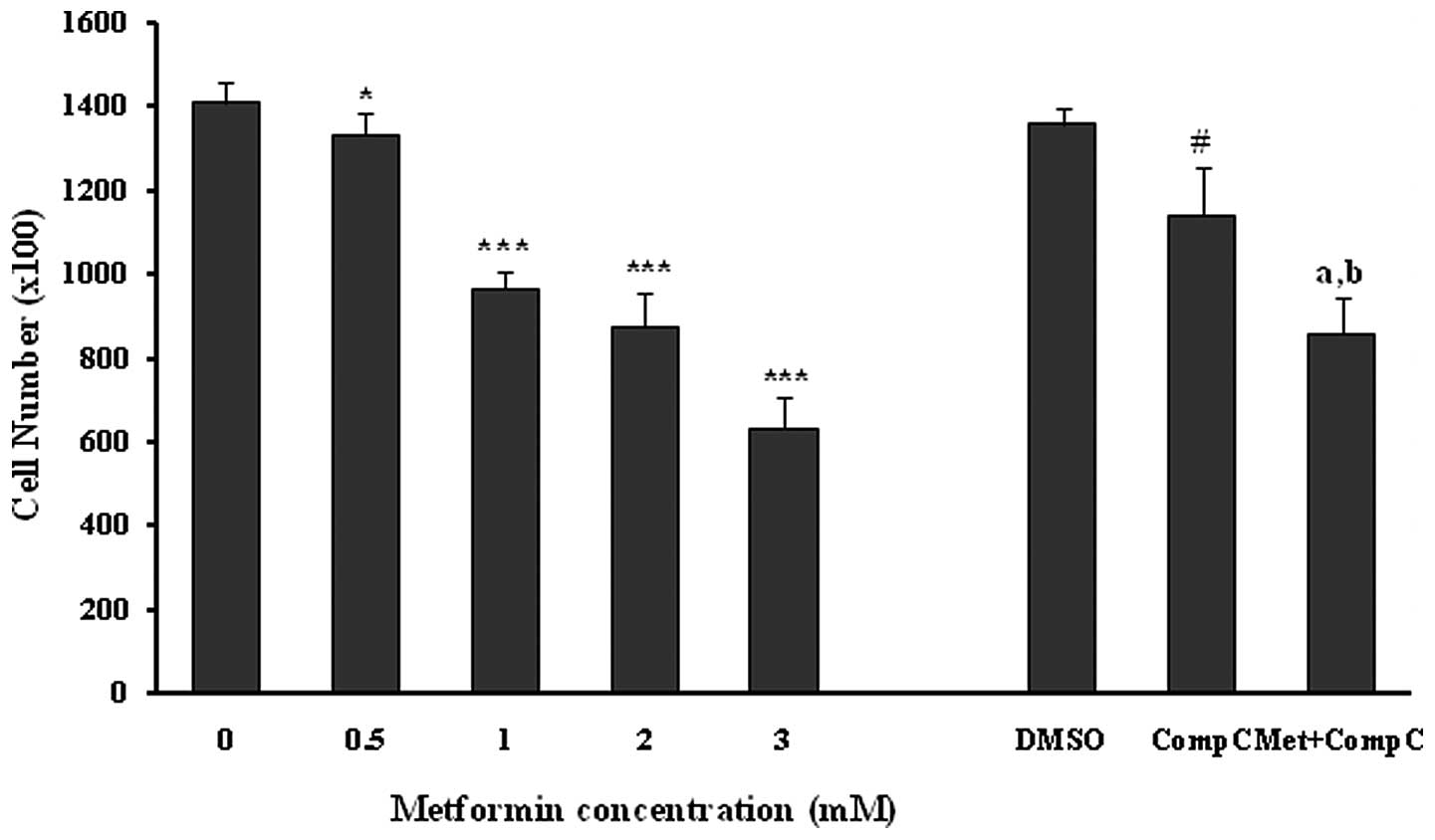
The anti-proliferative effects of metformin were confirmed using an MTT proliferation assay (Fig. 2), with similar significant (P<0.001) and concentration-dependent decreases noted in endothelial cell proliferation. However, compound C did not affect the cell viability in this set of experiments (Fig. 2). Metformin at 3 mM produced a significant (P<0.001; 35%) inhibition of endothelial cell proliferation in the presence of compound C. However, the inhibitory effect was much lower in comparison to the cells treated with metformin (3 mM) alone (P<0.001; 52%). The results of the MTT assay also showed that compound C partially blocked the anti-proliferative action of metformin (Fig. 2), and this was comparable with that of the cell counting experiments (Fig. 1).
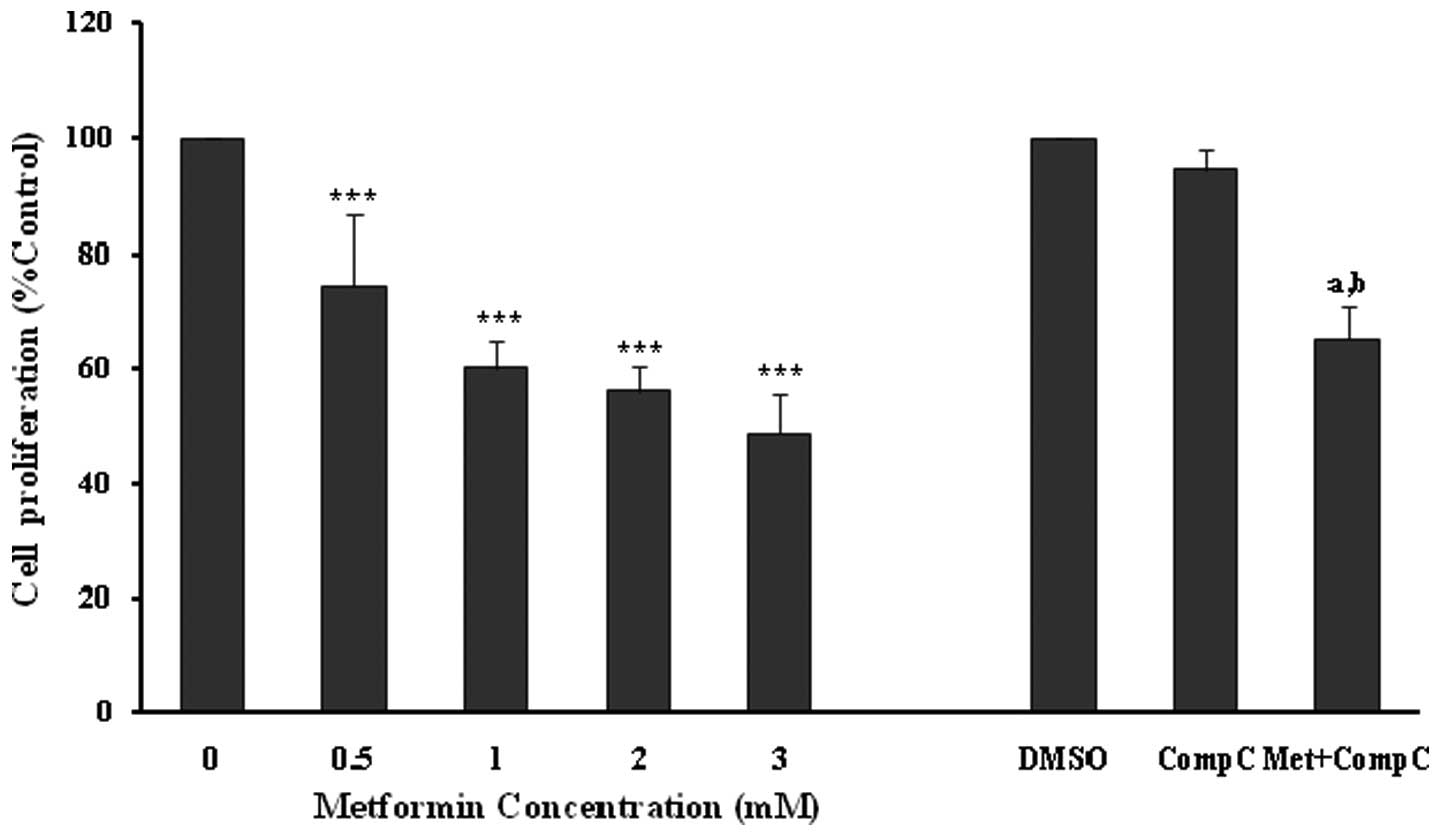
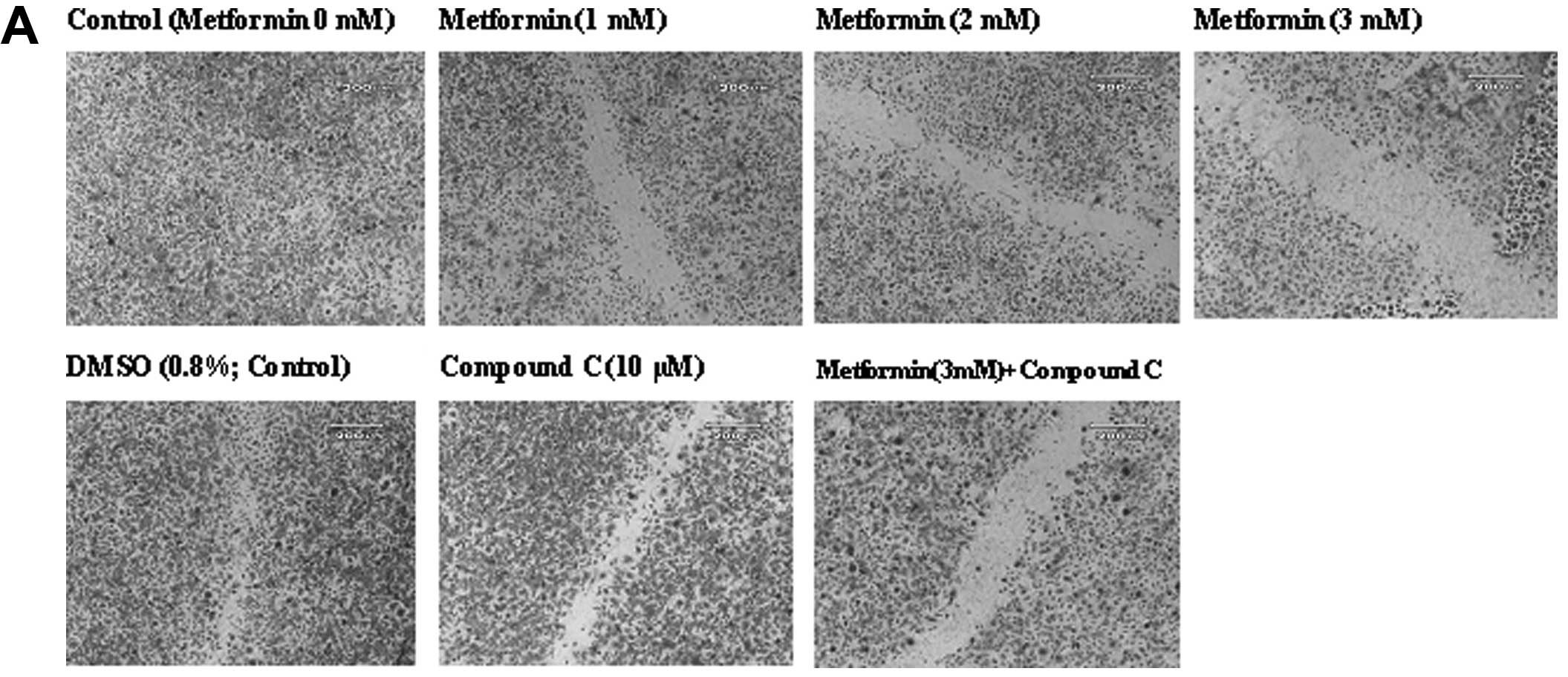
Effects of metformin on endothelial cell migration
The ‘wound’ repair model of migration was used to evaluate the effect of metformin on endothelial cell migration. Confluent scrape-wounded HUVEC monolayers were incubated for 72 h with metformin in the presence or absence of compound C. Subsequently, the degree of closure of the ‘wound’ was assessed. It was observed that metformin at concentrations of 0.5–3.0 mM induced a strong and significant (P<0.001) concentration-dependent inhibition of ‘wound’ repair in HUVECs from 31 to 80%, respectively (Fig. 3). Compound C significantly inhibited the migration (P<0.001), which was consistent with its effect on endothelial cell numbers. However, in comparison to the metformin alone-treated cells (3 mM), compound C partially, but significantly (P<0.001) reversed the anti-migratory effect of metformin.
Effects of metformin on MMP-2 and -9 expression in HUVECs
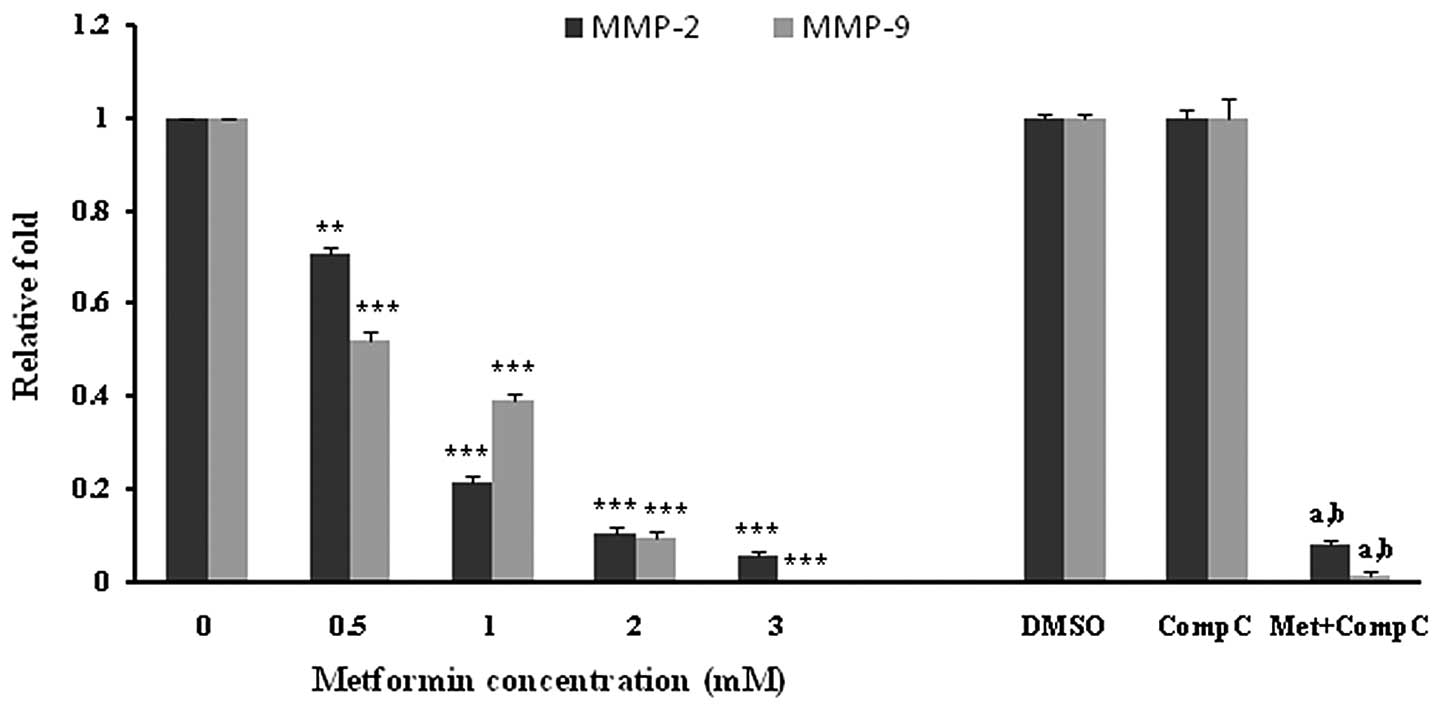
HUVECs were incubated with different concentrations of metformin for 72 h. Subsequently, the mRNA expression of MMP-2 and -9 was examined. Metformin significantly (P<0.001) decreased both the MMP-2 and -9 mRNA levels in a concentration-dependent manner (Fig. 4). The most marked decline in mRNA expression was noted with 3 mM of metformin. DMSO, as a vehicle, or compound C, as an AMPK inhibitor, had no effect on the mRNA expression of MMPs. However, it was observed that compound C significantly (P<0.05), but not completely, reversed the suppressive effect of metformin (3 mM) on the MMP-2 and -9 mRNA expression (Fig. 4).
Discussion
Angiogenesis is an integral part of tumor growth and metastasis that has gained increased interest as a core component in cancer therapy. Several case-control and observational cohort clinical trials have reported that systemic treatment with the anti-diabetic drug metformin considerably decreased the risk of different types of cancer in diabetic patients (1–3,18–20). A recent study on mice exposed to tobacco carcinogenesis demonstrated that metformin decreased the tumor burden by 72%, which was correlated with decreased cellular proliferation and marked inhibition of mTOR in tumors (21). However, it is necessary to clarify whether metformin exerts the anticancer effect, at least in part, through an anti-angiogenic effect. The present study found that metformin produced a potent anti-proliferative and anti-angiogenic effect in vitro on HUVECs, and that this effect was associated with reduced mRNA expression of MMP-2 and -9.
It was clear from the experiments that metformin produced a potent (P<0.001) and concentration-dependent inhibitory effect on HUVEC proliferation. A concentration of 3 mM of metformin reduced the endothelial cell numbers in culture by at least 55%. The effect was also confirmed by an MTT proliferation assay. The anti-proliferative effect of metformin was not due to cytotoxicity due to the fact that treatment of endothelial cells with different concentrations of metformin for extended periods of time neither affected the integrity of the cell monolayer nor was it associated with increased LDH release, an indicator of cytotoxicity. Furthermore, the obtained data showed that the migration of the cells into the denuded area 72 h after the cultures were treated with metformin was significantly (P<0.001) lower in comparison to the untreated control. To the best of our knowledge, not many studies have dealt with the effects of metformin on angiogenesis, in particular the effects of metformin on the endothelial cell proliferation and migration. A study conducted on PCOS women treated with metformin reported that metformin decreased angiogenesis by increasing the serum anti-angiogenic thrombospondin-1 (15). Also, a recent in vivo study in a murine sponge model demonstrated that metformin inhibited inflammatory angiogenesis by decreasing the levels of transforming growth factor-β1 (TGF-β1) (16). In addition, our findings revealed that the MMP-2 and -9 expression in unstimulated HUVECs was markedly downregulated, following the metformin treatment of endothelial cells in a concentration-dependent manner. Inhibitory action of metformin, as a pharmacological activator of AMPK, on the MMP expression has been described in human fibrosarcoma cells (22). However, no study has evaluated the effect of the drug on MMP expression in vascular endothelial cells. MMPs are involved in many endothelial cell processes, such as cell migration and angiogenesis, as well as in tumor invasion or metastasis. These enzymes play an important role in physiological tissue remodeling, and also in pathological remodeling associated with conditions such as wound healing and tumor growth. In particular, MMP-2 and -9, the two predominately expressed MMPs in endothelial cells, have been directly implicated in the process of endothelial cell migration. This is accomplished through proteolysis of the components of the extracellular matrix (13,14).
Metformin has been used as an anti-diabetic drug since 1957 (23). The drug reduces blood sugar levels mainly through three mechanisms: decreased hepatic glucose production (24,25), increased skeletal myocyte glucose uptake (26,27), and reduction of hepatic lipids (28). AMP-activated protein kinase (AMPK) provides a candidate target, which is capable of mediating the beneficial metabolic effects of metformin (4). AMPK is an important intracellular energy sensor, which activates the catabolic pathways that generate ATP. In addition, AMPK also inactivates ATP-consuming anabolic pathways when the cellular AMP/ATP ratio is increased (29). Proliferation and migration are ATP-consuming processes. Thus, AMPK activity may be required for optimal cell proliferation and survival, in particular under stress conditions. It was observed that tumor xenografts prepared from Ras-transformed mouse embryo fibroblasts lacking AMPK lose their ability to grow in a hypoxic environment (30), and 5-amino-4-imidazole carboxamide riboside (AICAR), an AMPK agonist, increases the angiogenesis of endothelial progenitor cells by phosphorylation of acetyl-coenzyme A carboxylase (ACC) and eNOS (31). Furthermore, it has been reported that the activation of AMPK signaling in endothelial cells is essential for angiogenesis under hypoxic conditions (32), but not in normoxia. On the contrary, a growing body of evidence indicates that AMPK activation inhibits the growth and/or survival of various cancer cell lines (33–38). It is now obvious that AMPK is regulated by a well-recognized tumor suppressor known as LKB1 (39), and that activation of AMPK by metformin requires LKB1. Furthermore, AMPK activation by metformin inhibits the mammalian target of rapamycin (mTOR), a protein that plays a critical role in transcription, cell growth, proliferation and migration (6,7).
It was observed that compound C, a cell-permeable pyrazolopyrimidine derivative, acts as a potent and selective ATP-competitive inhibitor of AMPK (4). In the present study, we demonstrated that the anti-proliferative and anti-migratory effects of metformin on endothelial cells as well as the inhibitory effect of metformin on mRNA expression of MMP-1 and MMP-2 were significantly but not completely blocked by compound C. This indicates that the AMPK pathway is involved, at least in part, in the anti-angiogenic action of metformin. Surprisingly, compound C alone showed a slight but significant anti-proliferative and anti-migratory action. These paradoxical effects in the present study probably imply the involvement of AMPK-dependent and AMPK-independent mechanisms in metformin anti-angiogenic actions. The other possibility is that both the activation and inhibition of AMPK cause anti-proliferative effects through different downstream pathways. Regarding the energy-saving and ATP-producing roles of AMPK through enhancing fatty acid oxidation, glycolysis and glucose uptake (30,40), especially in ATP deprivation conditions, it is conceivable that AMPK inhibition by compound C to some extent leads to the inhibitory effects on HUVEC proliferation and migration. However, AMPK activation has a wider role in reducing circulating levels of insulin-like growth factor and inhibition of cell differentiation, proliferation, and growth through the suppression of mTOR (41), elongation factor-2 (42), and the cyclin (43) pathways. In this study, comparison of the strong inhibitory effects of metformin, as an AMPK activator, with the weak suppressive effects of compound C, as an AMPK antagonist, on the proliferation and migration of human umbilical vein endothelial cells indicates the potentially beneficial effects of AMPK activation in preventing angiogenesis and related diseases.
Many factors have been identified as stimulators of the MMP expression in endothelial cells (44) but little is known about the inhibitors of these elements in normal cells under physiological conditions. Using an AMPKα-knockout mouse, Morizane et al (45) reported that total AMPKα deletion significantly elevated MMP-9 expression in embryonic fibroblast cells. The authors also demonstrated that AMPK activation by AICAR or by A769662 in wild-type fibroblasts suppressed MMP-9 expression. Thus, it was concluded that both the activity and the presence of AMPKα contribute as a regulator of MMP-9 expression. Similarly, the present study demonstrated that the expression of MMP-2 and -9 mRNA was decreased in HUVECs incubated with metformin and this decrease was reversed partially by compound C as an inhibitor of AMPK.
Collectively, the results of this study suggest that metformin may have potential effects in arresting the progression of tumors by inhibiting endothelial cell proliferation and migration through the suppression of MMP-2 and -9 mRNA expression. In addition, AMPK activity, at least in part, is required for the above-mentioned effects. In conclusion, the results may clarify the beneficial effect of metformin in reducing cancer incidence in diabetic patients receiving the drug.
Acknowledgements
The present study was supported by grants from the Research Vice Chancellors of the Tabriz University of Medical Sciences, Tabriz, Iran and from the Research Vice Chancellors of the Tehran University of Medical Sciences, Tehran, Iran.