Baicalein prevents human prion protein-induced neuronal cell death by regulating JNK activation
- Authors:
- Published online on: November 26, 2014 https://doi.org/10.3892/ijmm.2014.2010
- Pages: 439-445
Abstract
Introduction
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative diseases in humans and animals (1) that share a pathognomonic triad as a common histopathological trait: the spongiform vacuolation of grey matter, neuronal cell death, glial cell proliferation and, occasionally, amyloid deposition (2). One of the fundamental events related to TSE pathogenesis is the refolding of a host-encoded glycoprotein, the cellular prion protein (PrPC), into a protease-insensitive isoform (PrPSc) that aggregates into deposits of misfolded protein (3). Conversion into PrPSc is driven by the transition of a large N-terminal region of PrPC from a random coil to a β-sheet structure, which becomes predominant over the α-helix content (43 vs. 30%) (3). The synthetic human prion protein peptide (PrP) (106–126) corresponds to amino acid residues 106–126 of the prion protein placed in the flexibly disordered N-terminal domain. PrP (106–126) maintains some of the pathogenic and physiological properties of PrPSc, including the induction of the apoptosis of hippocampal neurons and the proliferation of astrocytes (4). Several biochemical and biological properties of PrPSc (β-sheet-rich structure, amyloidogenesis, and neurotoxic and gliotrophic effects) have been discovered (4–7) from an analysis of the biological activity of different amino acid segments, a peptide encompassing amino acids 106–126 of the PrP sequence [PrP (106–126)] and its ability to reproduce in vitro. The relevance of the 106–126 sequence in PrP biological activity was further demonstrated by the observation that this fragment contains the amyloidogenic palindrome AGAAAAGA sequence (amino acids 113–120), which is the most conserved region within PrP molecules of different species (8).
The treatment of neurotoxic and neurodegenerative diseases involves limiting reactive oxygen species (ROS) production and oxidative stress. A prevoius study addressed neurodegeneration from a similar mechanistic standpoint based on ROS and oxidative stress (9). Low levels of ROS, such as superoxide anion (O2⨪), hydroxyl radical (HO•), and H2O2, serve as signaling molecules in various cellular pathways that lead to growth and survival. However, high levels of ROS induce cellular damage and death through the oxidation of lipids, proteins and DNA (10,11). Mitochondrial failure caused by the aggregation of misfolded proteins is a hallmark of neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease and prion diseases (12–14). PrP (106–126), a neurotoxic prion protein, induces mitochondrial dysfunction that results in neurotoxicity (15).
The dried root of Scutellaria baicalensis (S. baicalensis) Georgi has been used traditionally as a Chinese herbal medicine (Huang-qin). Flavonoids extracted from S. baicalensis are effective in treating the hypoxic brain, chemical neuronal cell damage and cognitive impairment (16,17). One of the flavanoids, baicalein, has been shown to have antioxidant activity (18), exert protective effects against the cytotoxicity induced by oxidative stress (19), as well as some beneficial effects on the cardiovascular system (20,21), among other biological activities (22–24). A recent study demonstrated that baicalein exerts neuroprotective effects in patients with Parkinson’s disease (25).
In the present study, investigated the effects of baicalein on human prion protein-induced neuroblastoma cell apoptosis, as well as the cellular responses to prion protein-related apoptotic signaling, ROS and mitochondrial dysfunction. We also examined whether the inhibition of c-Jun N-terminal kinase (JNK) reduces the neuronal apoptosis caused by the prion peptide. Our results suggest that baicalein protects against PrP (106–126)-induced apoptosis, the production of ROS and mitochondrial dysfunction. Regulating the JNK pathway prevented neurotoxicity in neuroblastoma cells caused by the prion peptide.
Materials and methods
Cell culture
The human neuroblastoma cells (SH-SY5Y; SK-N-SH) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured in minimum essential medium (MEM; Invitrogen-Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen-Gibco), 100 U/ml penicillin and 0.1 mg/ml gentamycin in a humidified incubator maintained at 37°C and 5% CO2. The cells were treated for 1 h with baicalein (Sigma-Aldrich, St. Louis, MO, USA) and then exposed to 50 mM PrP (106–126) for 24 h with or without the JNK inhibitor, SP600125 (Sigma-Aldrich). Untreated cells were used as controls.
PrP (106–126) treatment
PrP (106–126) was synthesized as previously described (8). The synthetic PrP (106–126) peptide (sequence, Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly) was synthesized by Peptron (Seoul, Korea). The peptide was dissolved in sterile dimethyl sulfoxide at a stock concentration of 10 mM and stored at −4°C.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
TUNEL assay was carried out to measure the stage of cellular apoptosis by a TUNEL-based assay kit (BioVision, Mountain View, CA, USA). TUNEL analysis was performed according to the manufacturer’s instructions. Propidium iodide (PI) was employed to show the cell nuclei.
Annexin V assay
Apoptosis in the detached cells (SH-SY5Y and SK-N-SH) was assessed using an Annexin V assay kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s instructions. Annexin V levels were determined by measuring fluorescence at an excitation wavelength of 488 nm and an emission wavelength of 525/30 nm using a Guava easyCyte HT System (Millipore, Bedford, MA, USA).
DCFH-DA assay and visual detection
The SH-SY5Y cells were incubated in MEM containing 10 μM 2′,7′-dichlorodi-hydrofluorescein diacetate (H2-DCFDA) at 37°C for 30 min. The cells were washed with phosphate-buffer saline (PBS) and lysed in lysis buffer (25 mM HEPES; pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1 mM DTT and protease inhibitor mixture). The cells were transferred to a clear 96-well plate, and fluorescence emissions were measured at 515 nm (bottom read mode) with an excitation wavelength of 488 nm, using a SpectraMax® M2 (Molecular Devices, Sunnyvale, CA, USA). The SH-SY5Y cells were cultured on coverslips in a 24-well plate in MEM containing 10 μM H2-DCFDA at 37°C for 30 min. The cells were washed with PBS, mounted with DakoCytomation fluorescent medium (Dako, Carpinteria, CA, USA) and visualized using a fluorescence microscope (Nikon Eclipse 80i; Nikon Corporation, Tokyo, Japan).
Mitochondrial transmembrane potential (MTP) assay
The change in MTP was evaluated using the cationic fluorescent indicator, JC-1 (Molecular Probes, Eugene, OR, USA), in which J-aggregates in intact mitochondria become fluorescent red with an emission at 583 nm, indicating high or normal MTP and fluorescent green with an emission at 525 nm, indicating low MTP when it remains in the monomeric form in the cytoplasm. The SH-SY5Y cells were incubated in MEM containing 10 μM JC-1 at 37°C for 15 min, washed with PBS, and then transferred to a clear 96-well plate. JC-1 aggregate fluorescence emissions were measured at 583 nm with an excitation wavelength of 488 nm, and JC-1 monomer fluorescence intensity was measured with excitation and emission wavelengths of 488 and 525 nm, respectively, using a Guava easyCyte HT System (Millipore). The SH-SY5Y cells were cultured on coverslips in a 24-well plate, incubated in MEM containing 10 μM JC-1 at 37°C for 15 min, and washed with PBS. Finally, the cells were mounted with DakoCytomation fluorescent medium (Dako, Carpinteria, CA, USA) and visualized under a fluorescence microscope (Nikon Eclipse 80i; Nikon Corporation).
Western blot analysis
The SH-SY5Y and SK-N-SH cells were lysed in lysis buffer {25 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1 mM dithiothreitol and a protease inhibitor mixture}, whole cell proteins were electrophoretically resolved by 10–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Immunoreactivity was detected through sequential incubation with primary antibodies, horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents provided with the West Save Gold Detection kit (AbFrontier Co., Ltd., Seoul, South Korea). The primary antibodies used for immunoblotting were anti-phospho-SAPK/JNK (Cat. no. 9255; Cell Signaling Technology, Danvers, MA, USA), anti-phospho-Akt (Cat. no. 2118-1; Epitomics, Burlingame, CA, USA) and anti-β-actin (A5441; Sigma-Aldrich). Images were examined using a Fusion FX7 imaging system (Vilber Lourmat, Torcy Z.I. Sud, France). The densitometry of the signal bands was analyzed using Bio-1D software (Vilber Lourmat).
Statistical analysis
The unpaired t-test or Welch’s correction was used for comparison between the two groups. For multiple comparison, the one-way ANOVA followed by the Tukey-Kramer test was used. All statistical analyses were performed using GraphPad Prism software. Results were considered significant for values P<0.05, P<0.01 or P<0.001.
Results
Baicalein inhibits PrP (106–126)-induced neuronal apoptosis
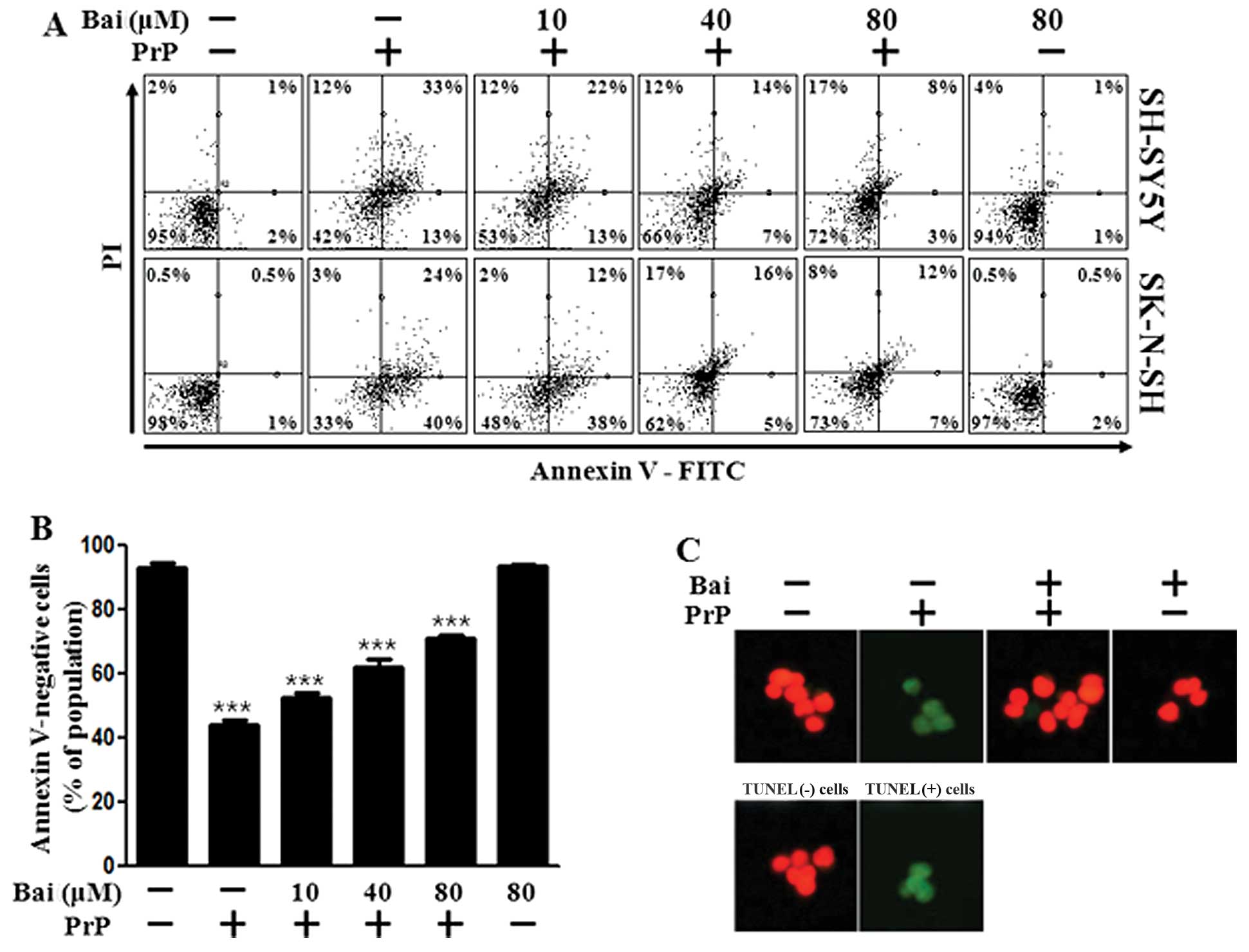
Baicalein has been shown to exert neuroprotective effects against neuronal impairment (26). In this study, we examined whether baicalein protects neuronal cells against prion-mediated neurotoxicity. Initially, we investigated the effects of baicalein on PrP (106–126)-induced neurotoxicity in SH-SY5Y and SK-N-SH cells using an Annexin V assay. The SH-SY5Y and SK-N-SH cells were exposed to baicalein with or without PrP (106–126). The cell viability of the PrP (106–126)-treated cells decreased by approximately 42% compared with that of the control SH-SY5Y cells and by 33% compared with that of the control SK-N-SH cells. The viability of the cells treated with baicalein only was similar to that of the untreated controls. Baicalein treatment inhibited the PrP (106–126)-induced neurotoxicity of the SH-SY5Y and SK-N-SH cells (Fig. 1A and B). We used a TUNEL assay (Fig. 1C) and microscopic imaging (Fig. 1D) and demonstrated that the apoptotic process in the PrP (106–126)-treated cells led to the emission of green fluorescence, indicating DNA strand breakage. Baicalein treatment recovered the cell death induced by PrP (106–126) treatment as measured by cell density (Fig. 1D). These results indicate that baicalein is effective in preventing PrP (106–126)-induced apoptosis in SH-SY5Y and SK-N-SH cells.
Treatment with baicalein exerts inhibitory effects on PrP (106–126)-mediated ROS production and mitochondrial dysfunction
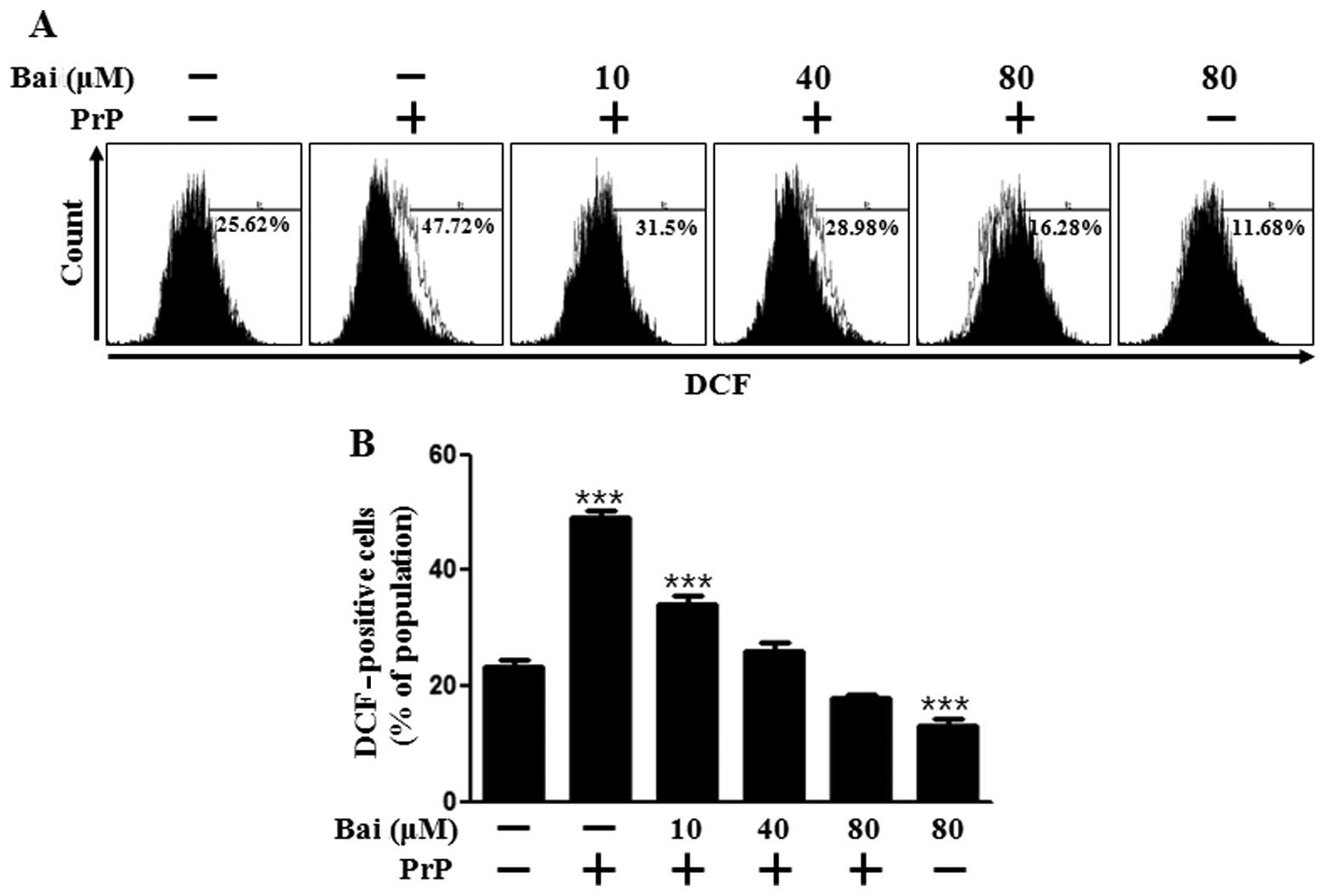
Oxidative stress is responsible for the neuronal cell death associated with prion diseases. Therefore, in this study, we investigated the mechanisms through which baicalein induces resistance to PrP (106–126) by assessing the antioxidant properties and generation of ROS in neuronal cells following tretment with baicalein. As shown in Fig. 2A and B, DCFH-DA fluorescence increased following treatment with PrP (106–126), whereas it was inhibited by baicalein treatment in a dose-dependent manner. We also assessed whether the protective effects of baicalein on PrP (106–126)-induced neurotoxicity are related to the prevention of mitochondrial dysfunction. The SH-SY5Y cells were pre-incubated with 10 μm baicalein for 12 h and then exposed to 50 μm PrP (106–126). The PrP (106–126)-treated cells displayed increased JC-1 monomers (58.2%), indicating low MTP values, whereas baicalein treatment reduced PrP (106–126)-induced JC-1 monomers (24.3%) in a dose-dependent manner, indicating high MTP values (Fig. 2C). In accordance with these results, the fluorescence microscopy images (Fig. 2D) showed cells with green fluorescence (JC-1 monomer form) following treatment with PrP (106–126), indicating lower MTP, whereas the control cells and baicalein-treated cells displayed red fluorescence (JC-1 aggregate form), indicating high MTP values. These data demonstrate that baicalein reduces ROS production and attenuates mitochondrial dysfunction induced by PrP.
Baicalein mediates survival and death signals
We then investigated whether baicalein exerts an effect on cell growth and death signals. Akt promotes growth factor-mediated cell survival both directly and indirectly. JNK is activated by PrP (106–126)-induced apoptosis (27). In addition, baicalein attenuates astroglial activation by downregulating the activation of JNK (28). Therefore, in this study, we examined Akt and JNK activation in neuronal cells following treatment with baicalein and PrP (106–126). Our results demonstrated that baicalein reversed the inhibition of Akt activation induced by treatment with PrP (106–126) and blocked JNK activation which was induced by PrP (106–126) (Fig. 3). These results suggest that baicalein exerts a protective intracellular signal. It can thus be hypothesized that the protective effects of baicalein may be inhibited by an Akt inhibitor. This suggests that baicalein plays a role similar to that of a JNK inhibitor under apoptotic conditions. Taken together, our results suggest that treatment with baicalein inhibits the PrP (106–126)-induced apoptosis of neuronal cells by regulating JNK. Therefore, this was examined in the following experiment.
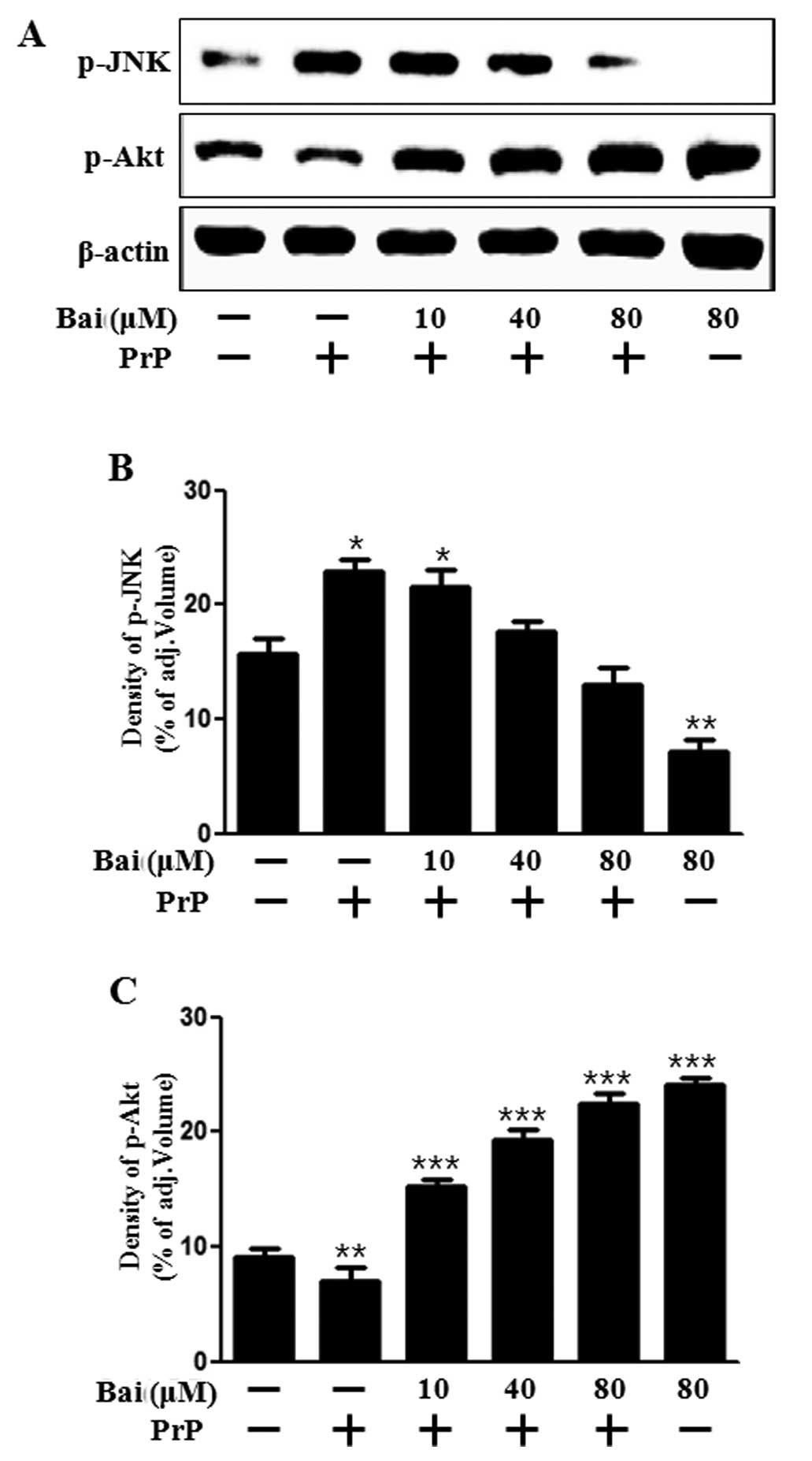
Baicalein inhibits PrP (106–126)-induced apoptosis by inactivating JNK
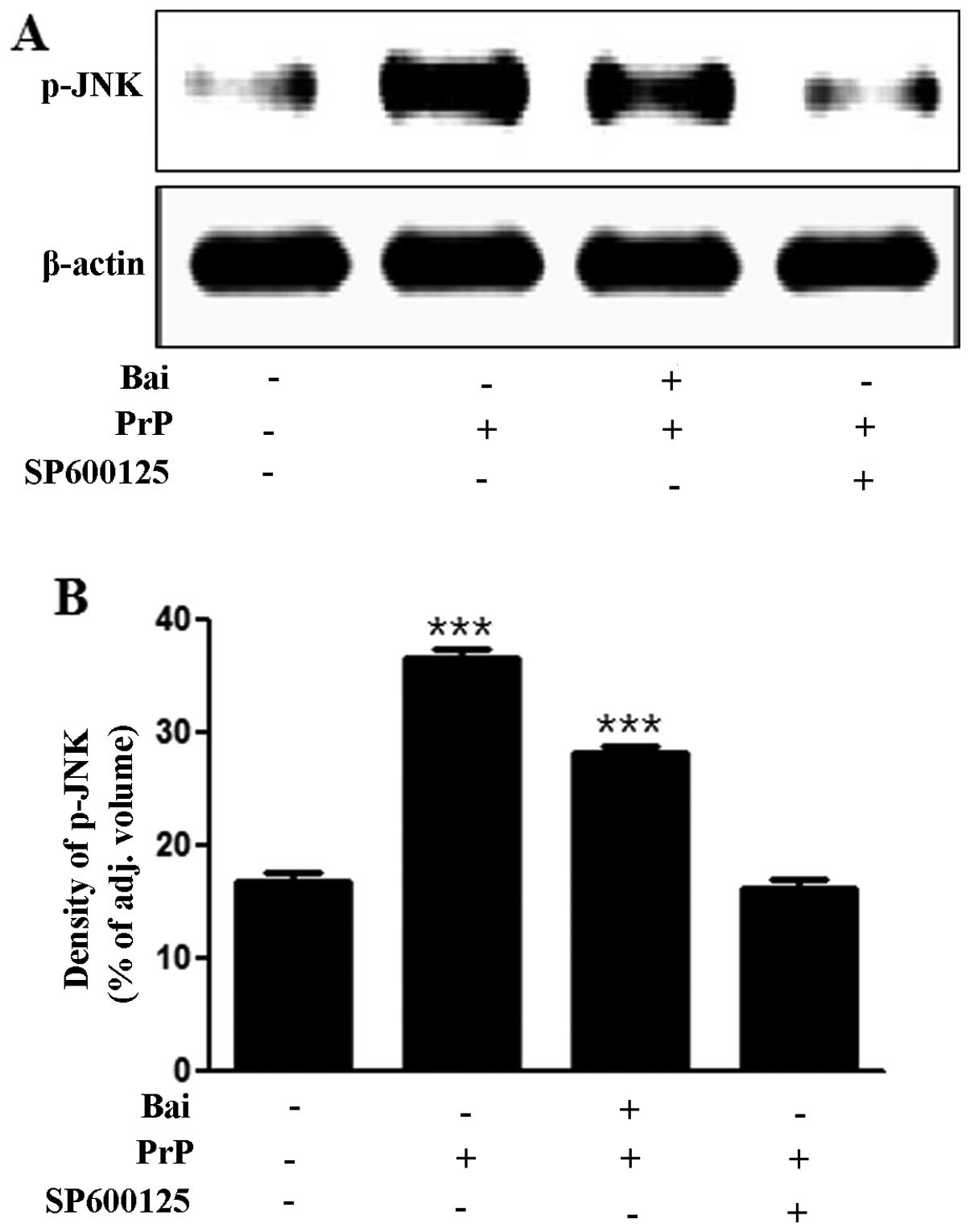
To investigate whether the inhibition of JNK exerts neuroprotective effects against PrP (106–126)-induced neurotoxicity, we analyzed the levels of JNK and cell viability using a JNK inhibitor (SP600125). We found that the PrP (106–126)-induced increase in JNK levels was inhibited by the JNK inhibitor, SP600125, as shown by western blot and densitometric analyses (Fig. 4A and B). We also found that the JNK inhibitor (SP600125) blocked the PrP (106–126)-induced neuronal apoptosis by inactivating JNK (Fig. 4C and D). The cells in the first quadrant of the graph in Fig. 4C signify the late apoptotic condition and those in the fourth quadrant signify the early apoptotic condition. These results strongly suggest that treatment with baicalein exerts neuroprotective effects against neuronal cell apoptosis by inactivating JNK.
Discussion
Baicalein, a flavonoid extracted from the traditional Chinese herbal medicine, S. baicalensis Georgi (Huang-qin), has been studied as a chemopreventive agent in neuronal cell and brain models (28–30). In the present study, we demonstrated that baicalein protected neuronal cells against PrP (106–126)-induced neurotoxicity.
Certain studies have suggested that high levels of ROS induce tissue injury, increase the formation of superoxide and induce neurotoxicity (31,32). As shown in the present study, treatment with baicalein attenuated the PrP (106–126)-induced production of ROS and neurotoxicity in dose-dependent manner (Figs. 1 and 2). These results indicate that ROS are associated with neurodegeneration and that baicalein protects against neurotoxicity induced by ROS. However, we were unable to demonstrate whether the inhibition of ROS production protects against PrP (106–126)-induced neurotoxicity. Further research on ROS and neurodegeneration is clearly required.
The mitochondria are the main source of cellular reactive oxygen, whose production further increases if the mitochondria are damaged (33,34). The contribution of ROS-induced mitochondrial dysfunction to the neurodegenerative process may depend on the cell type involved (35). As the mitochondria are both the main producers and targets of ROS, targeting mitochondrial oxidative damage may be clinically useful in neurodegeneration. Our data demonstrated that PrP (106–126) induced mitochondrial dysfunction and that baicalein protected the neuronal cells against mitochondrial damage (Fig. 2C and D). Our results are fragmentary, but suggest that baicalein protects neuronal cells against mitochondrial dysfunction and neurotoxicity from ROS overproduction induced by PrP.
In addition, we determined whether baicalein affects the mechanisms related to Akt and JNK signaling. Liu et al (30) suggested that the neuroprotective effects of baicalein involve the Akt pathway. Choi et al (36) proposed that baicalein protects hippocampal neuronal cells against apoptosis by inhibiting JNK activation. As expected, the JNK inhibitor, SP600125, protected the SK-N-SH cells against apoptotic death (Fig. 4C). However, Akt inhibitors (LY294002 and wortmannin) did not prevent the neuroprotective effects of baicalein (data not shown). Akt and JNK signals have been associated with baicalein, and our results indicate that JNK plays a key role regulating PrP (106–126)-induced neuronal apoptotic signals.
Several studies have suggested that JNK is associated with the generation of ROS. Shen et al (37) demonstrated that the JNK signal is a key modulator in cell death induced by ROS. It has also been suggested that the ROS-mediated JNK activation is a critical component deciding the fate of cells in response to various stressful stimuli, which can be inhibited by pre-treatment with antioxidants (38,39). Thus, further research on the association between JNK and ROS is required.
In conclusion, our results indicate that baicalein protects neuronal cells against PrP (106–126)-induced neurotoxicity by inhibiting JNK activation. In addition, baicalein reduces PrP (106–126)-induced mitochondrial dysfunction and ROS production. These data may prove useful to future studies and suggest the potential use of baicalein in the treatment of neurodegenerative disorders.
Acknowledgments
The present study was supported by a grant from the National Research Foundation of Korea (NRF), funded by the Korean government (2013R1A2A2A01009614).
References
|
Peretz D, Williamson RA, Kaneko K, et al: Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 412:739–743. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Aguzzi A: Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem. 97:1726–1739. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Caughey B, Raymond GJ, Ernst D and Race RE: N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol. 65:6597–6603. 1991.PubMed/NCBI | |
|
Selvaggini C, De Gioia L, Cantu L, et al: Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem Biophys Res Commun. 194:1380–1386. 1993. View Article : Google Scholar : PubMed/NCBI | |
|
Forloni G, Angeretti N, Chiesa R, et al: Neurotoxicity of a prion protein fragment. Nature. 362:543–546. 1993. View Article : Google Scholar : PubMed/NCBI | |
|
Florio T, Grimaldi M, Scorziello A, et al: Intracellular calcium rise through L-type calcium channels, as molecular mechanism for prion protein fragment 106–126-induced astroglial proliferation. Biochem Biophys Res Commun. 228:397–405. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
De Gioia L, Selvaggini C, Ghibaudi E, et al: Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J Biol Chem. 269:7859–7862. 1994.PubMed/NCBI | |
|
Corsaro A, Thellung S, Villa V, et al: Prion protein fragment 106–126 induces a p38 MAP kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells independently from the amyloid fibril formation. Ann N Y Acad Sci. 1010:610–622. 2003. View Article : Google Scholar | |
|
Halliwell B: Oxidative stress and neurodegeneration: where are we now? J Neurochem. 97:1634–1658. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Jabs T: Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol. 57:231–245. 1999. View Article : Google Scholar : PubMed/NCBI | |
|
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L and Elazar Z: Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Borger E, Aitken L, Muirhead KE, et al: Mitochondrial β-amyloid in Alzheimer’s disease. Biochem Soc Trans. 39:868–873. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Coskun P, Wyrembak J, Schriner SE, et al: A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 1820:553–564. 2012. View Article : Google Scholar : | |
|
Siskova Z, Mahad DJ, Pudney C, et al: Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am J Pathol. 177:1411–1421. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
O’Donovan CN, Tobin D and Cotter TG: Prion protein fragment PrP-(106–126) induces apoptosis via mitochondrial disruption in human neuronal SH-SY5Y cells. J Biol Chem. 276:43516–43523. 2001. View Article : Google Scholar | |
|
Shang YZ, Gong MY, Zhou XX, Li ST and Wang BY: Improving effects of SSF on memory deficits and pathological changes of neural and immunological systems in senescent mice. Acta Pharmacol Sin. 22:1078–1083. 2001.PubMed/NCBI | |
|
Shang Y, Cheng J, Qi J and Miao H: Scutellaria flavonoid reduced memory dysfunction and neuronal injury caused by permanent global ischemia in rats. Pharmacol Biochem Behav. 82:67–73. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Bochorakova H, Paulova H, Slanina J, Musil P and Taborska E: Main flavonoids in the root of Scutellaria baicalensis cultivated in Europe and their comparative antiradical properties. Phytother Res. 17:640–644. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Heo HJ, Kim DO, Choi SJ, Shin DH and Lee CY: Potent inhibitory effect of flavonoids in Scutellaria baicalensis on amyloid beta protein-induced neurotoxicity. J Agric Food Chem. 52:4128–4132. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Huang Y, Wong CM, Lau CW, et al: Inhibition of nitric oxide/cyclic GMP-mediated relaxation by purified flavonoids, baicalin and baicalein, in rat aortic rings. Biochem Pharmacol. 67:787–794. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Chen YC, Shen SC, Chen LG, Lee TJ and Yang LL: Wogonin, baicalin, and baicalein inhibition of inducible nitric oxide synthase and cyclooxygenase-2 gene expressions induced by nitric oxide synthase inhibitors and lipopolysaccharide. Biochem Pharmacol. 61:1417–1427. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Chang WS, Lee YJ, Lu FJ and Chiang HC: Inhibitory effects of flavonoids on xanthine oxidase. Anticancer Res. 13:2165–2170. 1993.PubMed/NCBI | |
|
Kyo R, Nakahata N, Sakakibara I, Kubo M and Ohizumi Y: Baicalin and baicalein, constituents of an important medicinal plant, inhibit intracellular Ca2+ elevation by reducing phospholipase C activity in C6 rat glioma cells. J Pharm Pharmacol. 50:1179–1182. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Liao JF, Hung WY and Chen CF: Anxiolytic-like effects of baicalein and baicalin in the Vogel conflict test in mice. Eur J Pharmacol. 464:141–146. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Yu X, He G and Du G: Neuroprotective effect of baicalein in patients with Parkinson’s disease. Zhongguo Zhong Yao Za Zhi. 37:421–425. 2012.in Chinese. PubMed/NCBI | |
|
Mu X, He GR, Yuan X, Li XX and Du GH: Baicalein protects the brain against neuron impairments induced by MPTP in C57BL/6 mice. Pharmacol Biochem Behav. 98:286–291. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Carimalo J, Cronier S, Petit G, et al: Activation of the JNK-c-Jun pathway during the early phase of neuronal apoptosis induced by PrP106–126 and prion infection. Eur J Neurosci. 21:2311–2319. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Lee E, Park HR, Ji ST, Lee Y and Lee J: Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1,2,3,4-tetrahydro-pyridine-induced Parkinson’s disease model by downregulating the activations of nuclear factor-kappaB, ERK, and JNK. J Neurosci Res. 92:130–139. 2014. View Article : Google Scholar | |
|
Lee JH and Lee SR: The effect of baicalein on hippocampal neuronal damage and metalloproteinase activity following transient global cerebral ischaemia. Phytother Res. 26:1614–1619. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Liu C, Wu J, Xu K, et al: Neuroprotection by baicalein in ischemic brain injury involves PTEN/AKT pathway. J Neurochem. 112:1500–1512. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Kovacic P and Somanathan R: Redox processes in neurode-generative disease involving reactive oxygen species. Curr Neuropharmacol. 10:289–302. 2012. View Article : Google Scholar : | |
|
Chakraborty S, Bornhorst J, Nguyen TT and Aschner M: Oxidative stress mechanisms underlying Parkinson’s disease-associated neurodegeneration in C. elegans. Int J Mol Sci. 14:23103–23128. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Beal MF: Mitochondria take center stage in aging and neurode-generation. Ann Neurol. 58:495–505. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Radi R, Cassina A and Hodara R: Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 383:401–409. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Bolanos JP, Moro MA, Lizasoain I and Almeida A: Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: therapeutic implications. Adv Drug Deliv Rev. 61:1299–1315. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Choi JH, Choi AY, Yoon H, et al: Baicalein protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis through inhibition of reactive oxygen species production and CHOP induction. Exp Mol Med. 42:811–822. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Shen HM and Liu ZG: JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 40:928–939. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Sato M, Bagchi D, Tosaki A and Das DK: Grape seed proanthocyanidin reduces cardiomyocyte apoptosis by inhibiting ischemia/reperfusion-induced activation of JNK-1 and C-JUN. Free Radic Biol Med. 31:729–737. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Yoshizumi M, Kogame T, Suzaki Y, et al: Ebselen attenuates oxidative stress-induced apoptosis via the inhibition of the c-Jun N-terminal kinase and activator protein-1 signalling pathway in PC12 cells. Br J Pharmacol. 136:1023–1032. 2002. View Article : Google Scholar : PubMed/NCBI |