
Role of autophagy and ferroptosis in the development of endometriotic cysts (Review)
- Authors:
- Published online on: July 11, 2024 https://doi.org/10.3892/ijmm.2024.5402
- Article Number: 78
-
Copyright: © Kobayashi et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Endometriosis is an estrogen-dependent condition affecting 10% of women in their reproductive life (1). It is characterized by the growth of endometrial-like tissues at sites outside of the uterus and is associated with pelvic pain and infertility. The pathogenesis of endometriosis is still unclear, but a number of factors are known, including retrograde menstruation, coelomic metaplasia, induction theory and hormonal, immunologic determinant, stem cell and genetic/epigenetic factors (2). According to Sampson's theory, endometriotic lesions are originated from the shed endometrial tissues of the uterus through retrograde menstrual dissemination, move and attach to the ectopic sites and then form endometriotic lesions (3). The retrograde endometrium encounters a number of challenges in the peritoneal cavity, including mechanical damage, abundant free iron and high oxidative levels, lack of oxygen, or immune attack (4). Periodic hemorrhage from ectopic endometriotic lesions is a hallmark of endometriosis and causes persistent iron overload (5). Despite a number of difficulties, endometriotic tissues may overcome these hurdles in the microenvironment by using mechanisms to escape from cell death. Retrograde menstruation occurs in as a ≥90% of menstruating women, but the incidence rate for endometriosis is only 10% (6). Thus, only a fraction of endometriotic cells may escape cell death in the harsh environment of intraperitoneal iron overload and hypoxia. This suggests that there must be other genetic and environmental factors that determine the onset and progression of this disease, such as mechanisms that prevent cell death (4).
Regulated cell death (RCD) is characterized by specific signaling cascades orchestrated by diverse biomolecules (7). RCD is further classified into apoptotic and non-apoptotic subcategories [i.e., autophagy (mitophagy), ferroptosis, necroptosis, pyroptosis, anoikis and cuproptosis] (7). Autophagy, defined as self-degradation, is a process related to nonapoptotic cell death induced by a large number of intracellular/extracellular stimuli (8). Damaged proteins and organelles are removed and recycled by autophagy (9). This process plays a pivotal role in maintaining quality control and cellular homeostasis. On the other hand, ferroptosis is defined as a reactive oxygen species (ROS)-dependent cell death related to iron accumulation and lipid peroxidation, which is different from other forms of cell death (10). We recently reported that autophagy is dynamically regulated by various intrinsic [e.g., phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mTOR) and extrinsic (e.g., hypoxia and oxidative stress)] pathways, effectively attenuating the induction of apoptosis and promoting the survival of endometriotic cells (11). The periodic and repeated bleeding in endometriotic lesions is thought to trigger iron-dependent cell death known as ferroptosis. However, endometriotic cells may have acquired a potential mechanism to escape cell death in the harsh environment of intraperitoneal iron overload, hypoxia and nutrient deprivation by regulating autophagy and ferroptosis. The present review summarized the current understanding of the mechanisms underlying autophagy and ferroptosis in endometriosis and discussed spatiotemporal orchestration of ferroptosis-mediated cell death regulation.
Methods
Search strategy and selection criteria
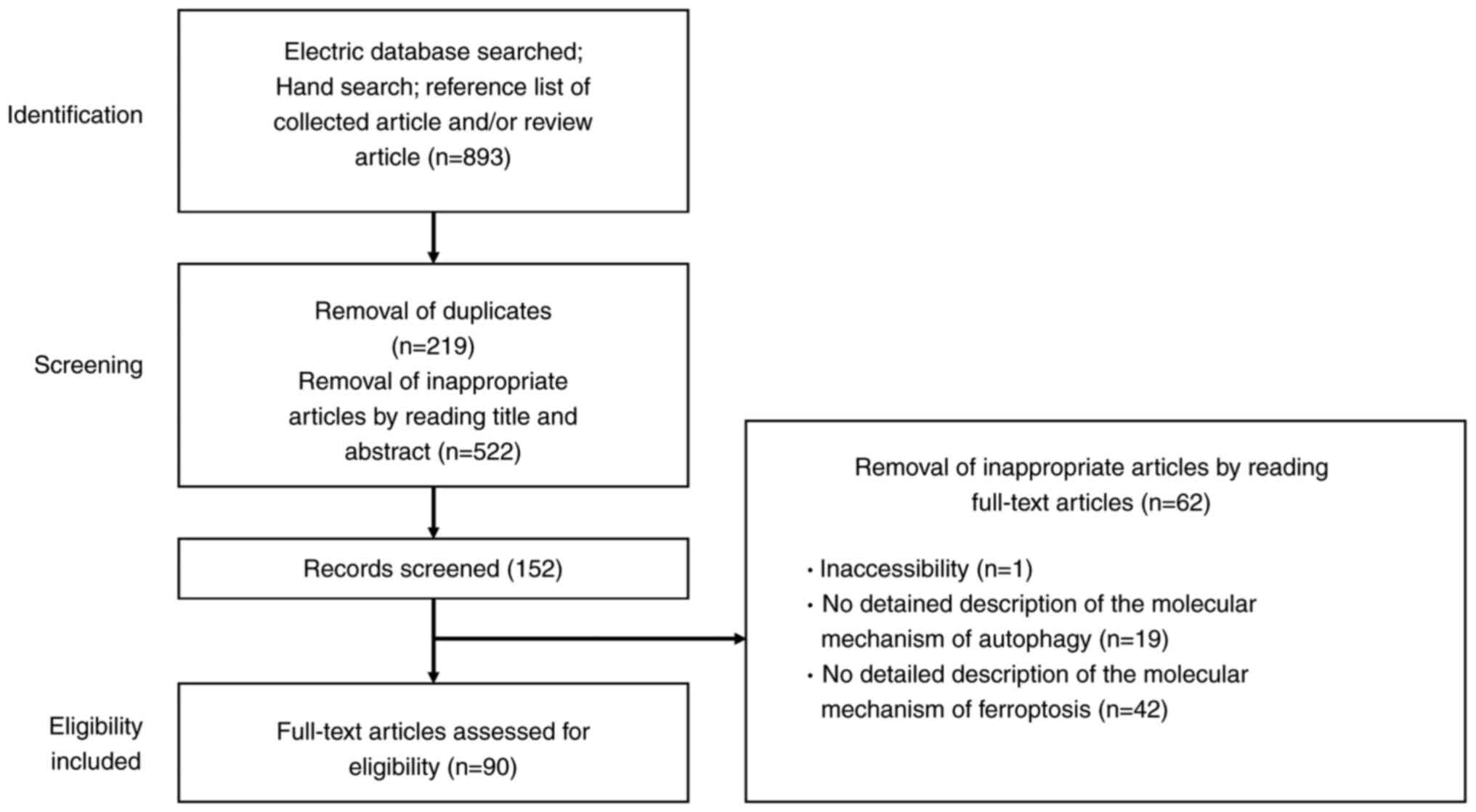
The present review conducted a narrative review of the targeted literature that focused on autophagy and ferroptosis in endometriosis. These mechanisms are well studied in cancer cells and the present review first drew information from these studies. It then summarized the roles of molecules regulating autophagy and ferroptosis that have been reported so far in endometriosis. Electronic databases including PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Google Scholar (https://scholar.google.jp/) were searched for literature published up to the October 31, 2023, combining the following keywords: 'Autophagy', 'ferroptosis', 'regulated cell death', 'survival', and 'endometriosis'. The search terms were combined using the Boolean operators And OR (Table I). Additionally, a manual reference search of published articles was conducted. Included studies comprised original research publications in English and reference lists from review articles. Duplicated studies, literature irrelevant to the research topic and non-English publications were excluded.
The flowchart depicted in Fig. 1 outlines the study selection process, detailing both inclusion and exclusion criteria. The initial phase involves identifying records through electronic database searches, manual searches and the reference lists of relevant articles and reviews. Titles and abstracts underwent a preliminary screening. After duplicates were removed, these titles and abstracts were reviewed to discard non-relevant studies. The final phase of eligibility involved analyzing the full-text articles, excluding any from which detailed data could not be obtained. The authors independently evaluated the articles to determine their suitability for inclusion or exclusion before reviewing the full texts. The properties of the identified molecules were searched in the National Library of Medicine database (https://www.ncbi.nlm.nih.gov/).
Molecular and regulatory network mechanisms controlling autophagy
This section briefly summarizes the current understanding of the development and progression of endometriosis, focusing on molecules that influence autophagy-mediated quality control. Autophagy is an evolutionarily conserved self-degrading process mediated by autophagosomes that eliminates various toxic substrates and maintains cellular homeostasis and survival (12). Autophagosomes, characterized by the formation of lipid-associated double membrane structures, fuse with lysosomes for cargo degradation (12,13). Mechanistically, autophagy is controlled by various autophagy-related gene (ATG) proteins (14). Among the ATG proteins, microtubule associated protein 1 light chain 3 (MAP1LC3, also known as LC3) and beclin1 (BECN1) have traditionally been evaluated as autophagy-related biomarkers (12,13). Beclin1 promotes autophagy and LC3 protein levels reflects autophagy induction (15). For more details, see our recent review article on the molecular mechanisms of autophagy and apoptosis in endometriosis (16). Autophagy can determine the cellular fate (14). For example, autophagy inhibits tumor initiation by eliminating potential oncogenic proteins and impaired organelles that cause genome instability (11,14). On the other hand, autophagy can maintain energy homeostasis and promote cancer cell growth by generating the necessary nutrients and energy during stress (11,14). Autophagy has the dichotomous role as both a tumor suppressor and tumor promoter. The present review summarized the role of autophagy in endometriosis, dividing it into pro-autophagy and anti-autophagy. Autophagy in endometriosis can be triggered by a number of factors such as oxidative stress, hypoxia, female hormones and specific signaling pathways (Fig. 2) (8).
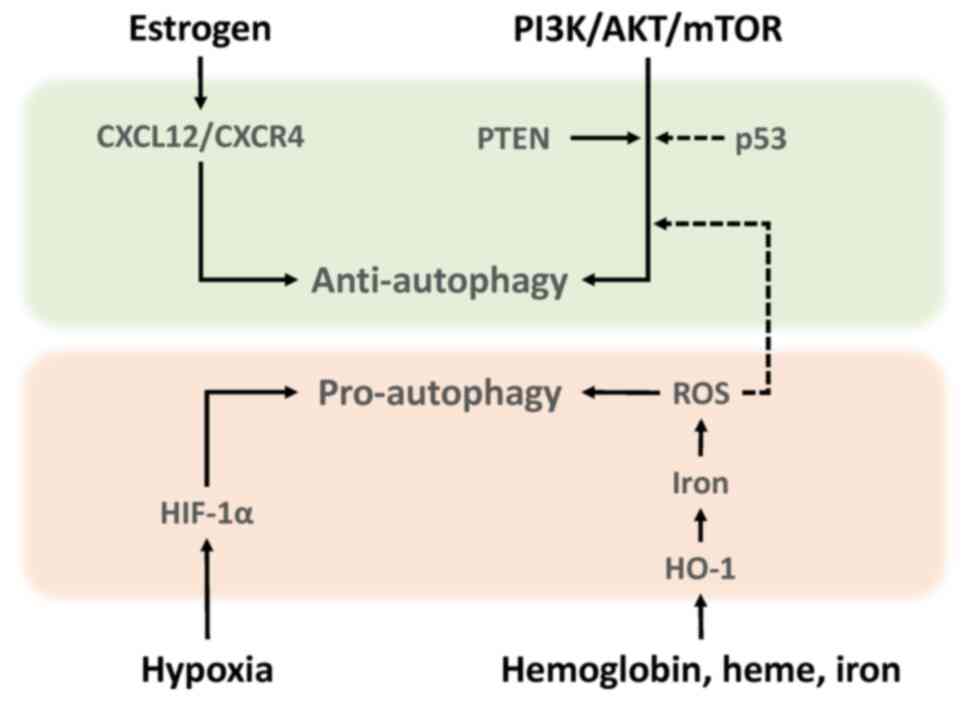
Anti-autophagy Estrogen
Estrogen promotes the proliferation of human endometrial stromal cells during the secretory phase via upregulation of C-X-C motif chemokine ligand 12 (CXCL12)/C-X-C motif chemokine receptor 4 (CXCR4) expression (17). CXCL12 has been reported to downregulate autophagy (17). It has been shown that levels of both CXCR4 and CXCL12 in endometriotic epithelial cells are higher than in normal endometrium (17,18). Therefore, estrogen dominance and progesterone resistance in endometriosis decrease autophagy levels through the activation of the CXCL12/CXCR4 axis (17,18). Estrogen-induced downregulation of autophagy is thought to promote the progression of endometriosis.
mTOR
mTOR expression level markedly increases in the order of normal endometrium, eutopic endometrium and ectopic endometrium (19,20). This suggests that the activation of the PI3K/AKT/mTOR pathway can reduce autophagy induction in endometriotic tissues. Notably, rapamycin, an mTOR inhibitor, confers therapeutic efficacy against endometriosis in animal models for endometriosis through upregulation of autophagy-related protein expression (13). Rapamycin also decreases cell viability and increases iron content and ROS generation (13). Additionally, phosphatase and tensin homolog (PTEN) and p53 are known to be genes involved in the regulation of autophagy in endometriosis. Activating mutations of PTEN gene are able to confer growth-promoting properties in endometriotic cells through regulation of autophagy induction (21). The frequency of PTEN gene mutations is ~21% in ovarian endometriosis (22). Tumor protein p53 (TP53), the most commonly mutated tumor suppressor gene, initiates cellular responses, such as cell cycle arrest, in response to stress signals (23). A reduction in p53 expression activates the mTOR pathway and negatively regulates autophagy, resulting in endometriosis cell proliferation (23). p53 expression was decreased in endometriotic tissues compared with normal endometria (24). Furthermore, there was a significant reduction in p53 expression in women with severe/late stage disease (25). Therefore, mTOR is considered a key gene that negatively regulates autophagy activity not only in cancer but also in endometriosis.
Pro-autophagy ROS
Iron-induced ROS induce autophagy through suppressing the mTOR signaling pathway (26). Moreover, hemoglobin, heme and iron caused by periodic hemorrhage induce HO-1 expression via ROS (27). HO-1 catalyzes heme breakdown to generate biliverdin, carbon monoxide and iron (27). HO-1 may further promote autophagy via iron overload (24).
Hypoxia
Additionally, hypoxia has been reported to induce enhancements of migration and invasion by activating HIF-1α-induced autophagy in human endometrial stromal cells (28,29). Indeed, HIF-1 promotes cell survival and blocks cell death in human ectopic endometriosis stromal cells (30).
From the aforementioned, both iron-mediated ROS and hypoxia-induced HIF-1α have been shown to induce autophagy in endometriotic cells. The generation of iron-mediated ROS that exceeds endogenous antioxidant capacity leads to oxidative cell damage. Therefore, well-coordinated quality control mechanisms via autophagy, involving timely removal of damaged multiple organelles, are essential for cellular homeostasis and survival. Considering the microenvironment, which is dominated by iron overload and hypoxia, effective autophagy could promote the very early stages of endometriosis development. Meanwhile, estrogen/CXCL12/CXCR4 23 (18) and PI3K/AKT/mTOR pathways (31) inhibit autophagy induction in endometriosis. Decreased autophagy may promote the growth and progression of pre-existing endometriotic lesions. Thus, both intrinsic and extrinsic factors have emerged as key regulators influencing autophagy (18,26,30,31). Collectively, autophagy may play a dual role in endometriosis cell survival, particularly in onset and progression. Previous reports have shown that autophagy is either activated or suppressed in endometriosis and some of the discrepancy between studies may greatly vary depending on environmental conditions and stage of disease (18,26,30,31).
Molecular and regulatory network mechanisms controlling iron metabolism and ferritinophagy
The development and progression of endometriosis may rely not only on the inherent proliferative nature but also on the ever-changing surrounding microenvironment (31,32). Endometriotic lesion growth can be promoted via multiple intrinsic pathways, including not only estrogen and PI3K/AKT/mTOR as aforementioned, but also mitogen-activated protein kinases (MAPK)/mitogen-activated protein kinase kinase/extracellular-signal-regulated kinases and nuclear factor-kappa B (33,34), while it may also be influenced by extrinsic factors such as iron overload and hypoxia (16,35,36). The present section mainly focused on the molecular mechanisms of iron, hypoxia-inducible factor 1 (HIF-1) and PI3K/AKT/mTOR pathways that regulate cell survival and summarized their roles in the development and progression of endometriosis.
Iron metabolism and its dynamics
Retrograde endometrial tissues encounter several challenges, including iron overload and hypoxia. Iron, ferritin and transferrin contents and transferrin saturation are markedly elevated in the peritoneal fluid of women with endometriosis compared with the controls (37). Moreover, an increase in follicular fluid iron and ferritin contents, transferrin saturation and transferrin receptor (TFRC) expression was demonstrated in the affected ovaries compared with the unaffected ovaries (38). Additionally, the content of iron is known to be ~1,000 times higher in the endometriotic cysts compared with other types of benign cysts (39). Endometriotic cysts contain large amounts of iron, but absolute concentrations vary by over 10-fold between patients (244.4±204.9 mg/l; range 65.3-1046.3 mg/l) (40). Furthermore, Iwabuchi et al (41) found that endometriotic cysts contain more ferric (Fe3+) than ferrous (Fe2+) ions. These findings have drawn much attention to the potential role of iron uptake, storage, detoxification, metabolism and iron homeostasis in the development, progression and pathogenesis of endometriosis (5). Circulating transferrin (TF) reversibly bound to Fe3+ is recognized by the TFRC and then transported to the endosome (Fig. 3) (42). After Fe3+ is reduced to Fe2+ by Steap3 metalloreductase within the endosomes, Fe2+ is transported out of endosomes via divalent metal transporter 1 (DMT1; also known as SLC11A2) (43). A portion of the cytosolic Fe2+ is transported to the mitochondria and the remaining labile iron is stored by ferritin, made up of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1) (42,43). Nuclear receptor coactivator 4 (NCOA4) is required for delivery of ferritin to lysosomes and its degradation (44). Thus, downregulation of NCOA4 inhibits ferritin degradation and reduces free iron level (44). Ferritinophagy is a specialized form of autophagy that is essential for transforming intracellular ferritin-bound iron into free iron. Finally, ferroportin (FPN; also known as SLC40A1) is the cellular iron exporter, maintaining cellular and systemic iron homeostasis (45). In addition, the labile iron pool causes the release of hazardous ROS (7) and oxidative stress (46) via Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH− + OH) (46). ROS include oxygen free radicals, such as superoxide anion, hydrogen peroxide, hydroxyl radical and free nitrogen radicals (27). On the other hand, endometriotic cells need increased antioxidant capacity to avoid cell death. Glutathione (GSH) and glutathione peroxidase (GPX) are known to play important roles as antioxidant defense systems that protect cells against oxidative damage in endometriosis (46). However, females with endometriosis show higher levels of oxidative stress markers [e.g., 8-hydroxy-2-deoxyguanosine, malondialdehyde (MDA), or lipid peroxide] and inadequate antioxidant defenses (e.g., superoxide dismutase, GPX4, HO-1 and catalase, GSH and vitamins A, C and E) compared with females without endometriosis (46,47). Therefore, endometriosis may avoid oxidative stress and maintain their viability in ways other than the antioxidant system. Indeed, an animal model showed that erythrocyte injection increased the number and size of endometriotic lesions relative to the untreated control groups (35). The use of desferrioxamine, an iron chelator, confirmed that iron was directly responsible for lesion growth (35). This seems to contradict an intuitive theory that iron induces cell death through oxidative damage. Iron may be essential for early lesion development. ROS is known to play a dual role as both beneficial (e.g., cell survival) and deleterious (e.g., cell death) effects (46), but the underlying mechanisms are unknown. The role of iron metabolism in the pathogenesis of endometriosis is a hot topic.
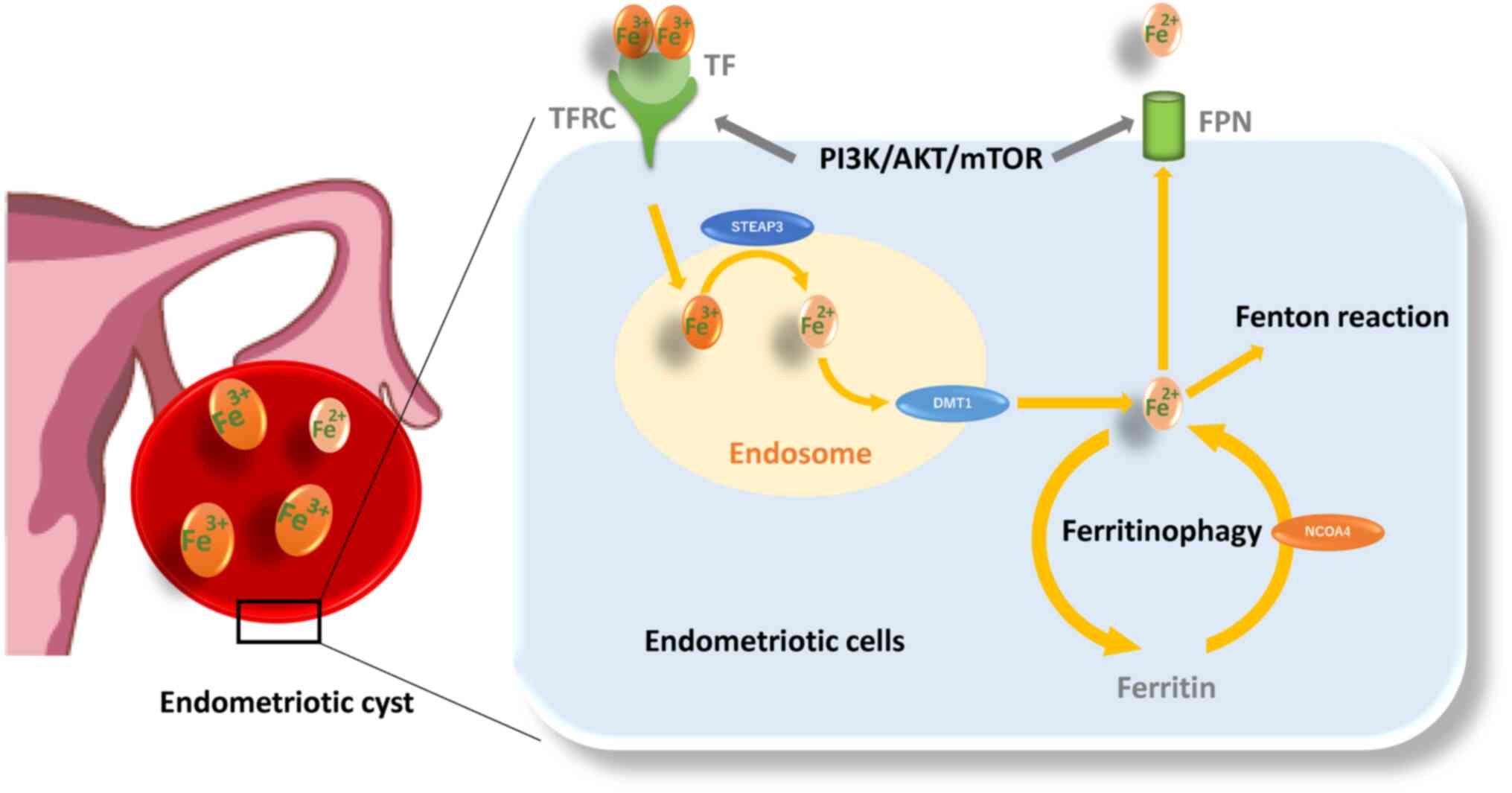
HIF-1 as a molecule regulating cell survival
In addition, hypoxia, a hallmark of endometriosis, plays a key role in the mitochondrial function and metabolic conversion between glycolysis and oxidative phosphorylation and metabolic remodeling is known to contribute to cell survival and disease progression (36). A hypoxic environment stimulates the proliferation of endometriotic cells through the upregulation of HIF-1α expression (48). Furthermore, HIF-1α regulates the expression of estrogen receptors, promotes inflammatory cytokine levels and accelerates angiogenesis (4). However, oxidative stress is also caused by hypoxia (48). Endometriotic cells being exposed to hypoxia-induced oxidative stress may acquire protective mechanisms to maintain their survival, avoiding cell cycle arrest or cell death from oxidative damage (48). Therefore, endometriotic lesion growth may be due to potential multiple factors that might affect the signaling mechanisms that control cell survival and death in response to iron overload and hypoxia.
Role of PI3K/AKT/mTOR pathway in regulating cell survival
The PI3K/AKT/mTOR axis is an important cell signaling pathway that is activated by steroid hormones and growth factors leading to cellular events (e.g., antioxidant, cell proliferation, differentiation, migration and survival) (33,49). mTOR regulates metabolism and energy homeostasis in response to nutrient signals, energy levels and stress stimuli with growth factors (31). Activation of the mTOR pathway upregulates the expression of TFRC and FPN, which reduces the net accumulation of intracellular iron, lowers the release of labile iron and regulates iron homeostasis (50). This pathway is known to be aberrantly activated in endometriosis (Fig. 2) (33,51). Therefore, activation of mTOR in endometriosis may suppress iron-mediated oxidative stress (50). These data suggest that in addition to the antioxidant defense systems, endometriotic cells may have evolved cellular mechanisms that control cell death to adapt to harsh environmental conditions such as iron overload and hypoxia.
Molecular and regulatory network mechanisms controlling ferroptosis
Ferroptosis is an iron-dependent non-apoptotic form of cell death through the accumulation of lipid peroxidation caused by ROS from the Fenton reaction (52). Ferroptosis can be triggered by iron overload and dysregulation of the transporter-dependent pathway [e.g., in a cystine/glutamate antiporter (xCT)-dependent manner] and/or the enzymic control mechanisms (e.g., in a GPX4-dependent manner) (53). The present section summarized the interrelationships of molecules and signaling pathways involved in ferroptosis. The essential role and pathways of each molecule are shown in Fig. 4.
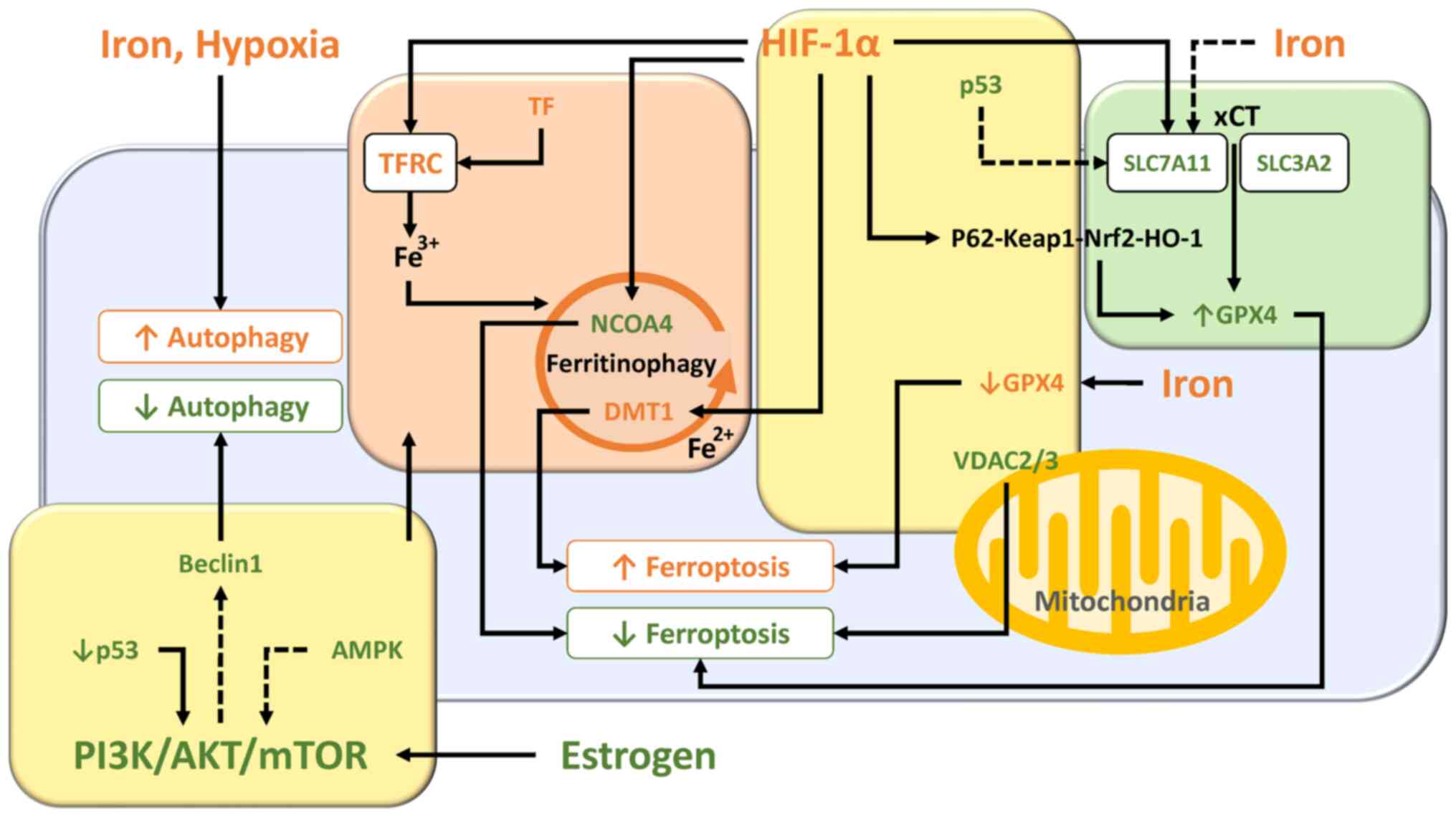
Molecular and regulatory network mechanisms controlled by iron overload (Fig. 4, orange box)
Differential gene expression analyses
Researchers have assessed the differences in ferroptosis-related genes in eutopic and ectopic endometrial samples from women with endometriosis and normal endometrium from women without endometriosis (54). Differential gene expression analyses based on the datasets in the Gene Expression Omnibus revealed that resistance to ferroptosis is a feature of eutopic and ectopic endometrial cells and changes of ferroptosis-related genes increased progressively from the eutopic endometrium to the ectopic endometrium (54). Increased SLC3A2 (a heavy-chain subunit of xCT) and TF expression and decreased NCOA4 and voltage-dependent anion channel 2/3 (VDAC2/3) expression were detected in eutopic endometrium compared with normal endometrium (54).
TF
Transferrin is an iron carrier protein that induces ferroptosis (55). Women with endometriosis have increased serum transferrin saturation levels as compared with controls (56). Furthermore, transferrin levels are elevated in the circulating and peritoneal fluid of females with endometriosis (57).
NCOA4
NCOA4-mediated ferritinophagy facilitates ferritin degradation in autolysosome, increases the cellular labile iron pool, triggers peroxidation of polyunsaturated fatty acid-containing phospholipids and activates ferroptosis induction (10,53,58). Accelerating the ferritinophagy system and downregulating ferroportin expression further increase susceptibility to ferroptosis (45). Decreased NCOA4 expression in endometriosis contributes to suppression of ferroptosis (54).
DMT1
Fe2+ is released into cytoplasm from the endosome via DMT1 (59). Iron is released into the cytoplasm by DMT1 overexpressed in the ectopic endometrium, induces oxidative stress via the Fenton reaction, leading to lipid peroxidation and ferroptosis (59).
Molecular and regulatory network mechanisms controlled by existing antioxidant systems (Fig. 4, green box)
xCT-GPX4
The xCT/GSH/GPX4 pathway is an important component for ferroptosis (60). A feature of ferroptosis is the reduction of antioxidant activity (e.g., intracellular GSH depletion and decreased activity of GPX4) (7). Excess iron decreases the expression of solute carrier family 7 member 11 (SLC7A11; a light-chain subunit of xCT) and GPX4, which efficiently increases susceptibility to ferroptosis (61). Blood injection results in GPX4 protein depletion in murine models with endometriosis iron overload (62). In fact, it has been reported that GPX4 expression in the early secretory phase is markedly lower in the eutopic endometrium of women with endometriosis compared with those with normal endometrium (63). These data suggest that endometriosis is more susceptible to iron-mediated ferroptosis.
Nuclear factor-E2-related factor 2 (Nrf2)
Nrf2, a key transcription factor that regulates cellular antioxidant response, increases the expression of genes involved in the anti-oxidative defense system, such as GPX4 (64). Nrf2 inhibits lipid peroxidation and ferroptosis via enhancing the antioxidant defense system. Protein expression of Nrf2 and its downstream molecules [e.g., NAD(P)H quinone dehydrogenase 1 (NQO1) and HO-1] in endometriotic lesions in a rat model showed an increased level compared with controls (65). Nrf2 has been known to be activated during autophagy (66). An adaptor protein Keap1 (kelch like ECH associated protein 1) binds Nrf2 and promotes ubiquitin-proteasome-mediated degradation of Nrf2 (65,67). Under oxidative stress conditions, phosphorylation and upregulation of p62, a selective autophagic cargo receptor, increases its binding affinity for Keap1, leading to Nrf2 dissociation from Keap1 (65-67). Free Nrf2 is transferred to the nucleus to induce the expression of various antioxidant genes and facilitate restoration of redox homeostasis (67). This suggests that Nrf2 is involved in ferroptosis resistance via activation of the autophagy process (66). Conversely, there is conflicting evidence that Nrf2 levels are lower in ectopic endometrium of endometriosis patients and in rat models of endometriosis compared with controls (20,68). Given that only activated Nrf2 that can translocate to the nucleus promotes ferroptosis resistance (67), intracellular total Nrf2 levels may be unrelated to ferroptosis. Therefore, quantification of Nrf2 mRNA and protein levels alone cannot determine the net effect of ferroptosis activation or inhibition.
HO-1
HO-1, a Nrf2-induced antioxidant enzyme, immediately removes toxic heme, catabolizes heme to biliverdin, carbon monoxide and iron and effectively protects cells from oxidative damage (41,69). For example, activation of the Keap1/Nrf2/HO-1 pathway may inhibit oxidative stress-mediated ferroptosis induced by lipopolysaccharide (LPS) (70,71). LPS is often used to induce oxidative stress and inflammation in animal models. On the other hand, HO-1 overexpression causes the accumulation of intracellular iron, promotes lipid peroxidation and triggers ferroptotic cell death (61,72,73). Therefore, HO-1 may exhibit a dual role as a suppressor or promoter of ferroptosis in response to oxidative stress, possibly depending on the degree of ferritinophagy and Nrf2 levels. HO-1 protein expression has been shown to be increased in ovarian endometriotic lesions (24), but its association with ferroptosis is still unclear.
Modulation of ferroptosis by molecules that control autophagy (Fig. 4, yellow box)
PI3K/Akt/mTOR
Certain oncogene mutations may play an important role in determining the susceptibility or resistance to ferroptosis (10). For example, aberrant constitutive activation of the PI3K/AKT/mTOR pathway markedly contributes to vital cellular functions, such as carcinogenesis, proliferation and survival and can acquire resistance to ferroptosis (10). Specific oncogenic driver mutations such as Kirsten rat sarcoma viral oncogene homologue and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α promoted ferroptosis resistance in lung cancer cells and breast cancer cells, respectively (10). Indeed, as illustrated in Fig. 3, the PI3K/Akt/mTOR pathway is known to influence iron metabolism and ferritinophagy.
AMPK
AMP-activated protein kinase (AMPK), a key cellular energy sensor, has been reported to either positively or negatively regulate ferroptosis (53). AMPK activated in response to energy stress and oxidative damage can inhibit ferroptosis via phosphorylation of downstream targets, including acetyl-CoA carboxylase α, the rate-limiting step in fatty acid synthesis (74) and Nrf2 (75). Meanwhile, the AMPK/Beclin1 signaling pathway promotes ferroptosis through inhibition of SLC7A11 activity in cancer cells (76). Beclin1 regulates not only autophagy but also ferroptosis (14). However, it remains unclear what role AMPK plays in ferroptosis, as there is conflicting evidence that expression of Beclin1 is increased (13,77) or decreased (78) in endometriosis.
VDAC
VDAC regulates flow of metabolites, ions and nucleotides across the outer membrane of the mitochondrion. A study suggests that VDAC is involved in autophagy, mitophagy, apoptosis and ferroptosis (79). VDAC ubiquitination orchestrated by a pathway involving the PTEN induced kinase 1 kinase and the parkin RBR E3 ubiquitin protein ligase (PARKIN) ubiquitin ligase eliminates damaged mitochondria by autophagy (80). Furthermore, dysregulation of mitochondrial function and overproduction of ROS by altering the permeability of VDAC promotes ferroptosis in lung cancer (81). Decreased VDAC2/3 expression in endometriosis may suppress ferroptosis (54).
HIF-1
Hypoxia is known to modulate ferroptosis primarily by upregulating HIF-1 expression (61). Hypoxia increases HIF-1-mediated iron uptake through upregulation of TFRC and DMT1 expression, promotes the accumulation of ROS and affects cell susceptibility to ferroptosis (61,82). In addition, the RNA-binding protein ELAV like RNA binding protein 1 interacts with HIF-1 mRNA, promotes its translation and triggers ferroptosis via activation of the ferritinophagy-mediated autophagy (61,83). Additionally, treatment of trophoblast cells with hypoxia reduces cell invasion and induces ferroptosis by downregulating SCL7A11, GPX4 and FPN1 expression (84). Furthermore, hypoxia/reoxygenation induces ferroptosis of cardiomyocytes (85). Meanwhile, hypoxia alleviates ferroptosis through decreasing NCOA4 levels, inhibiting ferritin degradation and increasing FTL and FTH1 storage in HT1080 fibrosarcome cells (86). Also, hypoxia reduces ferroptosis by inhibiting ferritinophagy in osteoclasts (87). Additionally, hypoxia/reoxygenation-induced oxidative stress may reduce ferroptosis through stearoyl-CoA desaturase 1-mediated fatty acid desaturation in cancer cells (88). Indeed, HIF-1 has been reported to inhibit ferroptosis in acute kidney injury (89). Hypoxia is also a central event in endometriosis. HIF-1α is known to promote eutopic endometrial stromal cell invasion by upregulating vascular endothelial growth factor and matrix metalloproteinases in endometriosis (29,90,91). Although HIF-1α has been reported to upregulate autophagy in endometriosis (29), it is unclear whether HIF-1α is involved in regulating ferroptosis. However, emerging evidence, such as decreased NCOA4 expression and increased Nrf2 and GPX4 expression in endometriosis, suggests that HIF-1α may regulate ferroptosis.
P53
p53 is known to have a dual role as a suppressor or promoter of ferroptosis (53,92). TP53 inhibits SLC7A11 expression to suppress tumor growth in human osteosarcoma, breast, or lung cancer cells through induction of GSH depletion and increased susceptibility to ferroptosis (93). TP53 triggers ferroptosis through enhanced arachidonate lipoxygenase expression and glutaminolysis by inducing the expression of spermidine/spermine N1-acetyltransferase 1 (94) and glutaminase 2 (55,92). Conversely, p53 inhibits ferroptosis through the upregulation of cyclin dependent kinase inhibitor 1 A/p21 (cyclin dependent kinase inhibitor 1 A) expression in cancer cells (95). Downregulation of p53 expression in endometriosis has been reported to suppress ferroptosis through activating the SLC7A11 pathway, facilitating disease progression (96).
The ferroptosis-related pathway in endometriosis
Based on these data, the present study summarized ferroptosis-related pathways in endometriosis (Fig. 4). First, it is well known that the SLC3A2/SLC7A11/Nrf2/GPX4 pathway controls ferroptosis (60). It has been reported that iron administration reduced GPX4 levels in rat models (early stages of endometriosis) (62). This result suggests the importance of ferroptosis induction for cell survival during the early stages of endometriosis development. Second, the gene expression levels of GPX4 in eutopic endometrium are higher than in the normal endometrium through upregulation of SLC3A2 expression (54). Upregulation of SLC3A2 (54) and SLC7A11 (96) expression suppresses ferroptosis and promotes disease progression. Furthermore, immunohistochemical studies showed that the positive rate of p53 in ectopic endometrium was lower than that in normal endometrium (97). Given that perturbations of the p53 locus occur frequently in severe/late stage endometriosis (25), downregulation of p53 expression may lead to the activation of the xCT-GPX4 pathway, promoting cell protection and survival by suppressing ferroptosis. Third, overexpression of TF enhances iron uptake in eutopic endometrium, but decreased NCOA4 expression inhibits ferritinophagy, promotes iron accumulation and suppresses Fe2+ release. In addition, increased expression of HO-1 and ferritin and decreased expression of DMT1 were detected in ectopic endometrium compared with eutopic endometrium. These data suggest that inhibition of ferritinophagy suppresses the accumulation of labile iron and ferroptosis (10,53,58,98,99). Finally, VDAC, which forms part of the mitochondrial permeability transition pore, is known to be involved in apoptotic cell death by promoting the release of cytochrome c (79). It has been reported that VDAC is closely involved in promoting ferroptosis in cancer cells (81,100). Similar to cancer cells, VDAC2/3 is markedly downregulated in the eutopic endometrium of patients with endometriosis (54). This indicates that decreased VDAC levels suppresses ferroptosis.
Alterations in autophagy and ferroptosis throughout the development of endometriotic cysts
Autophagy and ferroptosis have been shown to be induced or conversely suppressed in humans and preclinical animal models, but the reasons for such discrepancies and their mechanisms are still not fully understood. The present review discussed the role of autophagy and ferroptosis by dividing the development of endometriosis into four stages: Lesion initiation, induction of cell death, escape from cell death and formation of established lesions. Assuming that endometriosis evolves from retrograde endometrium, it was estimated how autophagy and ferroptosis change as endometriosis progresses (Fig. 5). Endometrial tissues originating from retrograde menstruation encounter environmental challenges, e.g., iron overload, oxidative stress and hypoxia caused by periodic bleeding (Fig. 5 ①) (4,46). Several lines of evidence indicate that iron has a crucial role in the proliferation of endometriotic cells. Ding et al (62) found that in comparison with control mice, endometriotic lesions in the iron-overload model mice were larger and more numerous. Under iron overload or hypoxic conditions, autophagy plays a positive role in scavenging ROS, preserving mitochondrial integrity, counteracting metabolic insults, avoiding apoptosis and protecting cells (10). Endometrial cells derived from retrograde menstruation may be able to survive even under harsh conditions, possibly through activation of autophagy-dependent quality control mechanisms (101). In endometriosis, ferroptosis can occur through two pathways, the activation of the autophagy pathway controlled by iron and HIF-1 (28,29) and the expression of ferritinophagy-related proteins such as DMT1 (59). Indeed, the levels of both iron and MDA, a secondary product of free radical lipid peroxidation, are increased in the peritoneal fluid and ovaries, whereas expression of anti-ferroptosis-related proteins (e.g., GPX4 and GSH) are decreased, highlighting the importance of ferroptosis (62,102). Moreover, increased ferroptosis is found to enhance the expression of pro-angiogenic factors vascular endothelial growth factor and IL-8 (103,104). Ferroptosis has been shown to contribute to development and maintenance of subsequent endometriotic lesions through modulating the interaction between inflammation and angiogenesis (101,103). Additionally, it has been reported that ferric ammonium citrate-induced ferroptosis may promote fibrosis process in endometriosis (102). Autophagy is essential for establishing early lesions by counteracting the ferroptosis-mediated deleterious effects. Only a rare fraction of cells are likely able to survive in harsh environments mediated by iron/hypoxia-dependent oxidative stress. It is easy to understand that an excessive ferroptosis induces the regression of endometriotic lesions via lipid peroxidation-driven membrane destruction (Fig. 5 ②). Indeed, erastin, a ferroptosis inducer, inhibits endometriotic lesion growth through iron accumulation and decreased FPN expression in a mouse model of endometriosis (45). Perhaps, excessive ROS induces autophagic cell death, accelerates iron-dependent ferroptosis and amplifies the process of lipid peroxidation and the extent of membrane rupture (105). Autophagy also plays a role in eliminating damaged proteins and subcellular organelles to sustain cell viability, while unrecoverable damage can trigger cell death (106). By contrast, autophagy removes damaged intracellular components, such as cellular proteins, lipids, nucleic acids and mitochondria and can inhibit ferroptosis (Fig. 5 ③). Nrf2-dependent autophagy activation may inhibit ferroptosis via enhancing the defense system against oxidative stress (65). Only a fraction of endometrial cells may receive survival signals and acquire a ferroptosis-resistant phenotype. On the other hand, intrinsic modulators (e.g., estrogen, PI3K/AKT/mTOR, AMPK, p53 and Beclin1) can inhibit autophagy through the expression of various ATG proteins (Fig. 5 ④). mTOR inhibitors have been reported to confer therapeutic efficacy against endometriosis through activation of autophagy (34). Furthermore, downregulation of the expression of NCOA4 and VDAC2/3 and upregulation of the expression of xCT/GPX4 induces the suppression of ferroptosis and causes further progression of endometriotic lesions.
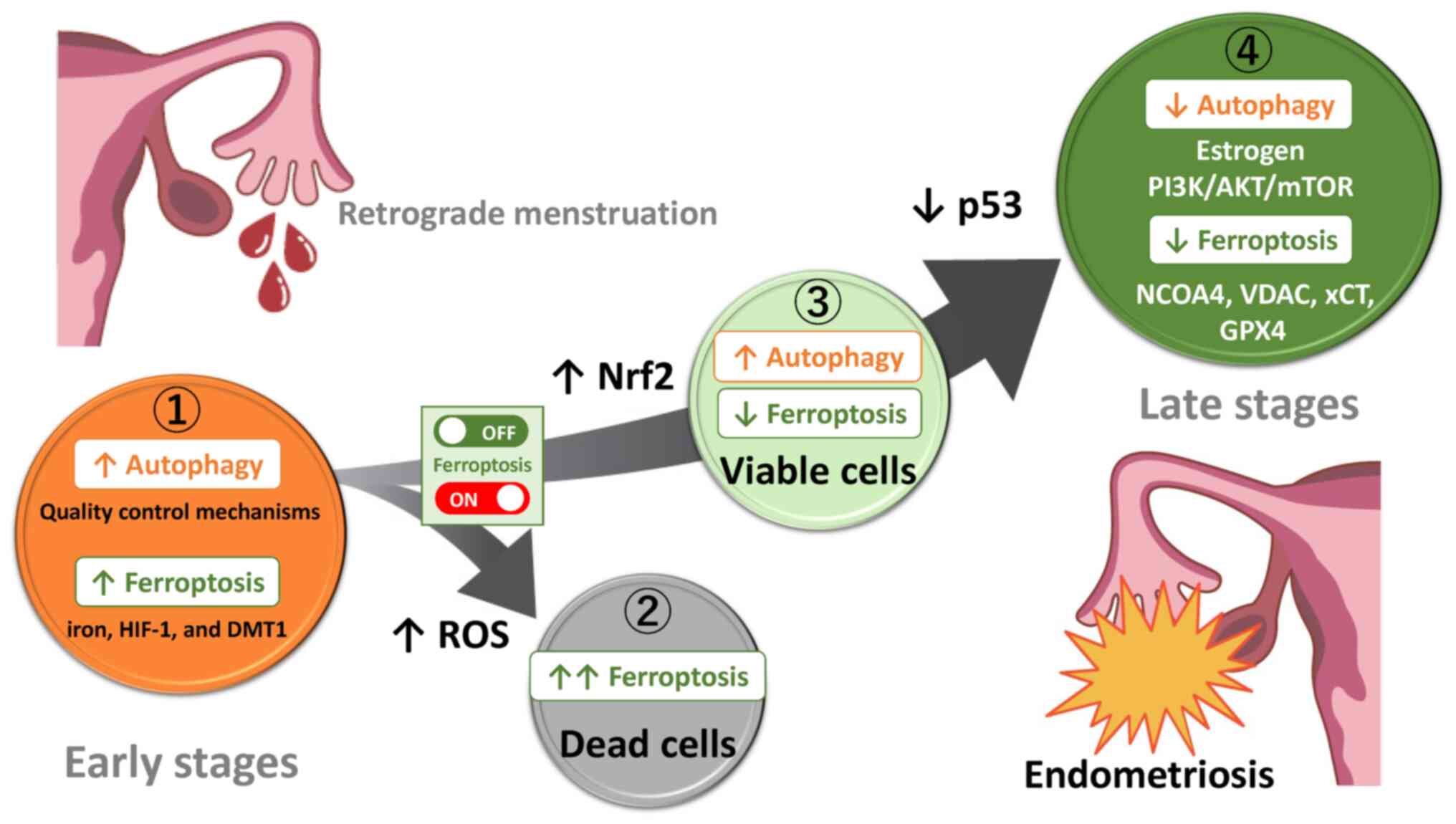
Conclusion
Induction of autophagy and ferroptosis may contribute to the survival and proliferation of the retrograde tissues originating from the endometrium, whereas suppression of autophagy and ferroptosis may play an important role in the progression of endometriotic lesions. The key molecular actors (e.g., estrogen, PI3K/AKT/mTOR, NCOA4, VDAC, xCT, GPX4, Nrf2, or p53) may switch ferroptosis on and off. Hence, in light of our prior report indicating that endometriotic cells may possess a potential anti-apoptotic capability to survive in changing environments (11), a timely fine-tuning of autophagy and ferroptosis levels could regulate the development and progression of endometriosis. In conclusion, autophagy and ferroptosis play a dual role in the initiation and progression of endometriosis through multiple mechanisms regulated by intrinsic factors (e.g., estrogen and the PI3K/AKT/mTOR pathway) or extrinsic stressors (e.g., iron overload, oxidative stress and hypoxia).
Availability of data and materials
Not applicable.
Authors' contributions
HK was responsible for conception and design of the present study. SM, CY and SI were responsible for acquisition of the data and HS was responsible for analysis and interpretation of data. HK drafted the manuscript and SM, CY, HS and SI performed a critical revision of the manuscript for important intellectual content. HS performed statistical analysis. H.K. provided administrative, technical or material support. SI supervised the present study. All authors read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Authors' information
Dr Hiroshi Kobayashi: ORCID: 0000-0002-8124-6269.
Abbreviations:
|
AKT |
protein kinase B |
|
AMPK |
AMP-activated protein kinase |
|
ATG |
autophagy-related gene |
|
BECN1 |
beclin1 |
|
CXCL12 |
C-X-C motif chemokine ligand 12 |
|
CXCR4 |
C-X-C motif chemokine receptor 4 |
|
DMT1 |
divalent metal transporter 1 |
|
FPN |
ferroportin |
|
FTL |
ferritin light chain |
|
FTH1 |
ferritin heavy chain 1 |
|
GPX |
glutathione peroxidase |
|
GSH |
glutathione |
|
HIF-1 |
hypoxia-inducible factor 1 |
|
HO-1 |
heme oxygenase-1 |
|
KEAP1 |
kelch like ECH associated protein 1 |
|
LC3 |
also known as MAP1LC3, microtubule associated protein 1 light chain 3 |
|
MAPK |
mitogen-activated protein kinases |
|
MDA |
malondialdehyde |
|
mTOR |
mammalian target of rapamycin |
|
NCOA4 |
nuclear receptor coactivator 4 |
|
Nrf2 |
Nuclear factor-E2-related factor 2 |
|
PI3K |
phosphatidylinositol 3-kinase |
|
PTEN |
phosphatase and tensin homolog |
|
RCD |
Regulated cell death |
|
ROS |
reactive oxygen species |
|
TF |
transferrin |
|
TFRC |
transferrin receptor |
|
TP53 |
tumor protein p53 |
|
VDAC |
voltage-dependent anion channel |
|
xCT |
cystine/glutamate antiporter |
Acknowledgements
Figures were created by Mrs. Toyomi Kobayashi (Ms.Clinic MayOne, Nara, Japan; https://www.mscl-mayone.com/; accessed on 9 January 31, 2024).
Funding
No funding was received.
References
|
Giudice LC: Clinical practice. Endometriosis. N Engl J Med. 362:2389–2398. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Vinatier D, Orazi G, Cosson M and Dufour P: Theories of endometriosis. Eur J Obstet Gynecol Reprod Biol. 96:21–34. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Sampson JA: Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. 14:422–469. 1927. View Article : Google Scholar | |
|
Li WN, Wu MH and Tsai SJ: Hypoxia and reproductive health: The role of hypoxia in the development and progression of endometriosis. Reproduction. 161:F19–F31. 2021. View Article : Google Scholar | |
|
Scutiero G, Iannone P, Bernardi G, Bonaccorsi G, Spadaro S, Volta CA, Greco P and Nappi L: Oxidative stress and endometriosis: A systematic review of the literature. Oxid Med Cell Longev. 2017:72652382017. View Article : Google Scholar : PubMed/NCBI | |
|
Viganò P, Parazzini F, Somigliana E and Vercellini P: Endometriosis: Epidemiology and aetiological factors. Best Pract Res Clin Obstet Gynaecol. 18:177–200. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Huang E, Wang X and Chen L: Regulated cell death in endometriosis. Biomolecules. 14:1422024. View Article : Google Scholar : PubMed/NCBI | |
|
Yang HL, Mei J, Chang KK, Zhou WJ, Huang LQ and Li MQ: Autophagy in endometriosis. Am J Transl Res. 9:4707–4725. 2017.PubMed/NCBI | |
|
Feng Y, He D, Yao Z and Klionsky DJ: The machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : | |
|
Lee S, Hwang N, Seok BG, Lee S, Lee SJ and Chung SW: Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis. 14:4642023. View Article : Google Scholar : PubMed/NCBI | |
|
Li X, He S and Ma B: Autophagy and autophagy-related proteins in cancer. Mol Cancer. 19:122020. View Article : Google Scholar : PubMed/NCBI | |
|
Glick D, Barth S and Macleod KF: Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Li H, Yang H, Lu S, Wang X, Shi X and Mao P: Autophagy-dependent ferroptosis is involved in the development of endometriosis. Gynecol Endocrinol. 39:22429622023. View Article : Google Scholar : PubMed/NCBI | |
|
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R and Tang D: Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem Biol. 27:420–435. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Kong Z and Yao T: Role for autophagy-related markers beclin-1 and LC3 in endometriosis. BMC Womens Health. 22:2642022. View Article : Google Scholar : PubMed/NCBI | |
|
Kobayashi H, Imanaka S, Yoshimoto C, Matsubara S and Shigetomi H: Molecular mechanism of autophagy and apoptosis in endometriosis: Current understanding and future research directions. Reprod Med Biol. 23:e125772024. View Article : Google Scholar : PubMed/NCBI | |
|
Mei J, Zhu XY, Jin LP, Duan ZL, Li DJ and Li MQ: Estrogen promotes the survival of human secretory phase endometrial stromal cells via CXCL12/CXCR4 up-regulation-mediated autophagy inhibition. Hum Reprod. 30:1677–1689. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Shen HH, Zhang T, Yang HL, Lai ZZ, Zhou WJ, Mei J, Shi JW, Zhu R, Xu FY, Li DJ, et al: Ovarian hormones-autophagy-immunity axis in menstruation and endometriosis. Theranostics. 11:3512–3526. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Guo J, Gao J, Yu X, Luo H, Xiong X and Huang O: Expression of DJ-1 and mTOR in eutopic and ectopic endometria of patients with endometriosis and adenomyosis. Gynecol Obstet Invest. 79:195–200. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Jamali N, Zal F, Mostafavi-Pour Z, Samare-Najaf M, Poordast T and Dehghanian A: Ameliorative effects of quercetin and metformin and their combination against experimental endometriosis in rats. Reprod Sci. 28:683–692. 2021. View Article : Google Scholar | |
|
Choi J, Jo M, Lee E, Hwang S and Choi D: Aberrant PTEN expression in response to progesterone reduces endometriotic stromal cell apoptosis. Reproduction. 153:11–21. 2017.PubMed/NCBI | |
|
Sato N, Tsunoda H, Nishida M, Morishita Y, Takimoto Y, Kubo T and Noguchi M: Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: Possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 60:7052–7056. 2000. | |
|
Cui D, Qu R, Liu D, Xiong X, Liang T and Zhao Y: The cross talk between p53 and mTOR pathways in response to physiological and genotoxic stresses. Front Cell Dev Biol. 9:7755072021.PubMed/NCBI | |
|
Allavena G, Carrarelli P, Del Bello B, Luisi S, Petraglia F and Maellaro E: Autophagy is upregulated in ovarian endometriosis: A possible interplay with p53 and heme oxygenase-1. Fertil Steril. 103:1244–1251.e1. 2015.PubMed/NCBI | |
|
Bischoff FZ, Heard M and Simpson JL: Somatic DNA alterations in endometriosis: High frequency of chromosome 17 and p53 loss in late-stage endometriosis. J Reprod Immunol. 55:49–64. 2002.PubMed/NCBI | |
|
Poillet-Perez L, Despouy G, Delage-Mourroux R and Boyer-Guittaut M: Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 4:184–192. 2015.PubMed/NCBI | |
|
Iwabuchi T, Yoshimoto C, Shigetomi H and Kobayashi H: Oxidative stress and antioxidant defense in endometriosis and its malignant transformation. Oxid Med Cell Longev. 2015:8485952015.PubMed/NCBI | |
|
Xu TX, Zhao SZ, Dong M and Yu XR: Hypoxia responsive miR-210 promotes cell survival and autophagy of endometriotic cells in hypoxia. Eur Rev Med Pharmacol Sci. 20:399–406. 2016.PubMed/NCBI | |
|
Liu H, Zhang Z, Xiong W, Zhang L, Xiong Y, Li N, He H, Du Y and Liu Y: Hypoxia-inducible factor-1alpha promotes endometrial stromal cells migration and invasion by upregulating autophagy in endometriosis. Reproduction. 153:809–820. 2017.PubMed/NCBI | |
|
Tsuzuki T, Okada H, Shindoh H, Shimoi K, Nishigaki A and Kanzaki H: Effects of the hypoxia-inducible factor-1 inhibitor echinomycin on vascular endothelial growth factor production and apoptosis in human ectopic endometriotic stromal cells. Gynecol Endocrinol. 32:323–328. 2016. | |
|
McKinnon BD, Kocbek V, Nirgianakis K, Bersinger NA and Mueller MD: Kinase signalling pathways in endometriosis: Potential targets for non-hormonal therapeutics. Hum Reprod Update. 22:382–403. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Wyatt J, Fernando SM, Powell SG, Hill CJ, Arshad I, Probert C, Ahmed S and Hapangama DK: The role of iron in the pathogenesis of endometriosis: A systematic review. Hum Reprod Open. 2023:hoad0332023. View Article : Google Scholar : PubMed/NCBI | |
|
Hung SW, Zhang R, Tan Z, Chung JPW, Zhang T and Wang CC: Pharmaceuticals targeting signaling pathways of endometriosis as potential new medical treatment: A review. Med Res Rev. 41:2489–2564. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang M, Xu T, Tong D, Li S, Yu X, Liu B, Jiang L and Liu K: Research advances in endometriosis-related signaling pathways: A review. Biomed Pharmacother. 164:1149092023. View Article : Google Scholar : PubMed/NCBI | |
|
Defrère S, Van Langendonckt A, Vaesen S, Jouret M, González Ramos R, Gonzalez D and Donnez J: Iron overload enhances epithelial cell proliferation in endometriotic lesions induced in a murine model. Hum Reprod. 21:2810–2816. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Kobayashi H, Shigetomi H and Imanaka S: Nonhormonal therapy for endometriosis based on energy metabolism regulation. Reprod Fertil. 2:C42–C57. 2021. View Article : Google Scholar | |
|
Defrère S, Lousse JC, González-Ramos R, Colette S, Donnez J and Van Langendonckt A: Potential involvement of iron in the pathogenesis of peritoneal endometriosis. Mol Hum Reprod. 14:377–385. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Sanchez AM, Papaleo E, Corti L, Santambrogio P, Levi S, Viganò P, Candiani M and Panina-Bordignon P: Iron availability is increased in individual human ovarian follicles in close proximity to an endometrioma compared with distal ones. Hum Reprod. 29:577–583. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Yamaguchi K, Mandai M, Toyokuni S, Hamanishi J, Higuchi T, Takakura K and Sujii S: Contents of endometriotic cysts, especially the high concentration of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin Cancer Res. 14:32–40. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Yoshimoto C, Iwabuchi T, Shigetomi H and Kobayashi H: Cyst fluid iron-related compounds as useful markers to distinguish malignant transformation from benign endometriotic cysts. Cancer Biomark. 15:493–499. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Iwabuchi T, Yoshimoto C, Shigetomi H and Kobayashi H: Cyst fluid hemoglobin species in endometriosis and its malignant transformation: The role of metallobiology. Oncol Lett. 11:3384–3388. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Gao G, Li J, Zhang Y and Chang YZ: Cellular iron metabolism and regulation. Adv Exp Med Biol. 1173:21–32. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Sendamarai AK, Ohgami RS, Fleming MD and Lawrence CM: Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc Natl Acad Sci USA. 105:7410–7415. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Li Y, Zeng X, Lu D, Yin M, Shan M and Gao Y: Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum Reprod. 36:951–964. 2021. View Article : Google Scholar | |
|
Kobayashi H, Yoshimoto C, Matsubara S, Shigetomi H and Imanaka S: Current understanding of and future directions for endometriosis-related infertility research with a focus on ferroptosis. Diagnostics (Basel). 13:19262023. View Article : Google Scholar : PubMed/NCBI | |
|
Matsuzaki S and Schubert B: Oxidative stress status in normal ovarian cortex surrounding ovarian endometriosis. Fertil Steril. 93:2431–2432. 2010. View Article : Google Scholar | |
|
Dai Y, Lin X, Xu W, Lin X, Huang Q, Shi L, Pan Y, Zhang Y, Zhu Y, Li C, et al: MiR-210-3p protects endometriotic cells from oxidative stress-induced cell cycle arrest by targeting BARD1. Cell Death Dis. 10:1442019. View Article : Google Scholar : PubMed/NCBI | |
|
Makker A, Goel MM, Das V and Agarwal A: PI3K-Akt-mTOR and MAPK signaling pathways in polycystic ovarian syndrome, uterine leiomyomas and endometriosis: An update. Gynecol Endocrinol. 28:175–181. 2012. View Article : Google Scholar | |
|
Bayeva M, Khechaduri A, Puig S, Chang HC, Patial S, Blackshear PJ and Ardehali H: mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. 16:645–657. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Driva TS, Schatz C and Haybaeck J: Endometriosis-associated ovarian carcinomas: How PI3K/AKT/mTOR pathway affects their pathogenesis. Biomolecules. 13:12532023. View Article : Google Scholar : PubMed/NCBI | |
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al: Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Tang D, Chen X, Kang R and Kroemer G: Ferroptosis: Molecular mechanisms and health implications. Cell Res. 31:107–125. 2021. View Article : Google Scholar : | |
|
Li B, Duan H, Wang S and Li Y: Ferroptosis resistance mechanisms in endometriosis for diagnostic model establishment. Reprod Biomed Online. 43:127–138. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Gao M, Monian P, Quadri N, Ramasamy R and Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Xu G, Chen L and Li Q: Association of iron metabolism markers, socioeconomic and lifestyle factors with endometriosis: A cross-sectional study. J Trace Elem Med Biol. 78:1271752023. View Article : Google Scholar : PubMed/NCBI | |
|
Mathur SP: Autoimmunity in endometriosis: Relevance to infertility. Am J Reprod Immunol. 44:89–95. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Santana-Codina N, Gikandi A and Mancias JD: The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol. 1301:41–57. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Alvarado-Díaz CP, Núñez MT, Devoto L and González-Ramos R: Endometrial expression and in vitro modulation of the iron transporter divalent metal transporter-1: Implications for endometriosis. Fertil Steril. 106:393–401. 2016. View Article : Google Scholar | |
|
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 171:273–285. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Gao X, Hu W, Qian D, Bai X, He H, Li L and Sun S: The mechanisms of ferroptosis under hypoxia. Cell Mol Neurobiol. 43:3329–3341. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Ding J, Zhao Q, Zhou Z, Cheng W, Sun S, Ni Z and Yu C: Huayu jiedu fang protects ovarian function in mouse with endometriosis iron overload by inhibiting ferroptosis. Evid Based Complement Alternat Med. 2022:14068202022. View Article : Google Scholar : PubMed/NCBI | |
|
Ota H, Igarashi S, Kato N and Tanaka T: Aberrant expression of glutathione peroxidase in eutopic and ectopic endometrium in endometriosis and adenomyosis. Fertil Steril. 74:313–318. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Dodson M, Castro-Portuguez R and Zhang DD: NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23:1011072019. View Article : Google Scholar : PubMed/NCBI | |
|
Kapoor R, Sirohi VK, Gupta K and Dwivedi A: Naringenin ameliorates progression of endometriosis by modulating Nrf2/Keap1/HO1 axis and inducing apoptosis in rats. J Nutr Biochem. 70:215–226. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 51:618–631. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Liu S, Pi J and Zhang Q: Signal amplification in the KEAP1-Nrf2-ARE antioxidant response pathway. Redox Biol. 54:1023892022. View Article : Google Scholar : PubMed/NCBI | |
|
Marcellin L, Santulli P, Chouzenoux S, Cerles O, Nicco C, Dousset B, Pallardy M, Kerdine-Römer S, Just PA, Chapron C and Batteux F: Alteration of Nrf2 and glutamate cysteine ligase expression contribute to lesions growth and fibrogenesis in ectopic endometriosis. Free Radic Biol Med. 110:1–10. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Loboda A, Damulewicz M, Pyza E, Jozkowicz A and Dulak J: Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol Life Sci. 73:3221–3247. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Li J, Lu K, Sun F, Tan S, Zhang X, Sheng W, Hao W, Liu M, Lv W and Han W: Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 19:962021. View Article : Google Scholar : PubMed/NCBI | |
|
Luo L, Huang F, Zhong S, Ding R, Su J and Li X: Astaxanthin attenuates ferroptosis via Keap1-Nrf2/HO-1 signaling pathways in LPS-induced acute lung injury. Life Sci. 311:1210912022. View Article : Google Scholar : PubMed/NCBI | |
|
Ryter SW: Heme oxgenase-1, a cardinal modulator of regulated cell death and inflammation. Cells. 10:5152021. View Article : Google Scholar : PubMed/NCBI | |
|
Machado SE, Spangler D, Stacks DA, Darley-Usmar V, Benavides GA, Xie M, Balla J and Zarjou A: Counteraction of myocardial ferritin heavy chain deficiency by heme oxygenase-1. Int J Mol Sci. 23:83002022. View Article : Google Scholar : PubMed/NCBI | |
|
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al: Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 22:225–234. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Z, Yao M, Jiang L, Wang L, Yang Y, Wang Q, Qian X, Zhao Y and Qian J: Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed Pharmacother. 154:1135722022. View Article : Google Scholar | |
|
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al: AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc-activity. Curr Biol. 28:2388–2399.e5. 2018. View Article : Google Scholar | |
|
Huang J, Chen X and Lv Y: HMGB1 mediated inflammation and autophagy contribute to endometriosis. Front Endocrinol (Lausanne). 12:6166962021. View Article : Google Scholar : PubMed/NCBI | |
|
Sui X, Li Y, Sun Y, Li C, Li X and Zhang G: Expression and significance of autophagy genes LC3, beclin1 and MMP-2 in endometriosis. Exp Ther Med. 16:1958–1962. 2018.PubMed/NCBI | |
|
Zhao Y, Li Y, Zhang R, Wang F, Wang T and Jiao Y: The role of erastin in ferroptosis and its prospects in cancer therapy. Onco Targets Ther. 13:5429–5441. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 12:119–131. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Vu NT, Kim M, Stephenson DJ, MacKnight HP and Chalfant CE: Ceramide kinase inhibition drives ferroptosis and sensitivity to cisplatin in mutant KRAS lung cancer by dysregulating VDAC-mediated mitochondria function. Mol Cancer Res. 20:1429–1442. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Xiong J, Nie M, Fu C, Chai X, Zhang Y, He L and Sun S: Hypoxia enhances HIF1 α transcription activity by upregulating KDM4A and mediating H3K9me3, thus inducing ferroptosis resistance in cervical cancer cells. Stem Cells Int. 2022:16088062022. View Article : Google Scholar | |
|
Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P and Zheng SY: ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. 27:142021. View Article : Google Scholar : PubMed/NCBI | |
|
Wang Y, Zhang L and Zhou X: Activation of Nrf2 signaling protects hypoxia-induced HTR-8/SVneo cells against ferroptosis. J Obstet Gynaecol Res. 47:3797–3806. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Liu XJ, Lv YF, Cui WZ, Li Y, Liu Y, Xue YT and Dong F: Icariin inhibits hypoxia/reoxygenation-induced ferroptosis of cardiomyocytes via regulation of the Nrf2/HO-1 signaling pathway. FEBS Open Bio. 11:2966–2976. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Fuhrmann DC, Mondorf A, Beifuß J, Jung M and Brüne B: Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36:1016702020. View Article : Google Scholar : PubMed/NCBI | |
|
Ni S, Yuan Y, Qian Z, Zhong Z, Lv T, Kuang Y and Yu B: Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic Biol Med. 169:271–282. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, Wery C, Collignon A, Longuespée R, Montero-Ruiz L, et al: Tumor resistance to ferroptosis driven by Stearoyl-CoA desaturase-1 (SCD1) in cancer cells and fatty acid biding protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 43:1020062021. View Article : Google Scholar : PubMed/NCBI | |
|
Li W, Xiang Z, Xing Y, Li S and Shi S: Mitochondria bridge HIF signaling and ferroptosis blockage in acute kidney injury. Cell Death Dis. 13:3082022. View Article : Google Scholar : PubMed/NCBI | |
|
Zhan L, Wang W, Zhang Y, Song E, Fan Y and Wei B: Hypoxia-inducible factor-1 alpha: A promising therapeutic target in endometriosis. Biochimie. 123:130–137. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Xiong W, Zhang L, Xiong Y, Liu H and Liu Y: Hypoxia promotes invasion of endometrial stromal cells via hypoxia-inducible factor 1α upregulation-mediated β-catenin activation in endometriosis. Reprod Sci. 23:531–541. 2016. View Article : Google Scholar | |
|
Kang R, Kroemer G and Tang D: The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 133:162–168. 2019. View Article : Google Scholar | |
|
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520:57–62. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Ou Y, Wang SJ, Li D, Chu B and Gu W: Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD and Dixon SJ: p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22:569–575. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Zou W, Wang X, Xia X, Zhang T, Nie M, Xiong J and Fang X: Resveratrol protected against the development of endometriosis by promoting ferroptosis through miR-21-3p/p53/SLC7A11 signaling pathway. Biochem Biophys Res Commun. 692:1493382024. View Article : Google Scholar | |
|
Sang L, Fang QJ and Zhao XB: A research on the protein expression of p53, p16, and MDM2 in endometriosis. Medicine (Baltimore). 98:e147762019. View Article : Google Scholar : PubMed/NCBI | |
|
Wang X, Zhou L, Dong Z and Wang G: Identification of iron metabolism-related predictive markers of endometriosis and endometriosis-relevant ovarian cancer. Medicine (Baltimore). 102:e334782023. View Article : Google Scholar : PubMed/NCBI | |
|
Goodall M and Thorburn A: Identifying specific receptors for cargo-mediated autophagy. Cell Res. 24:783–784. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Oh SJ, Ikeda M, Ide T, Hur KY and Lee MS: Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 8:4142022. View Article : Google Scholar : PubMed/NCBI | |
|
Ng SW, Norwitz SG, Taylor HS and Norwitz ER: Endometriosis: The role of iron overload and ferroptosis. Reprod Sci. 27:1383–1390. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Liu X, Deng M, Xu C, Zhang Y, Wu D, Tang F, Yang R and Miao J: Ferroptosis induced by iron overload promotes fibrosis in ovarian endometriosis and is related to subpopulations of endometrial stromal cells. Front Pharmacol. 13:9306142022. View Article : Google Scholar : PubMed/NCBI | |
|
Li G, Lin Y, Zhang Y, Gu N, Yang B, Shan S, Liu N, Ouyang J, Yang Y, Sun F and Xu H: Endometrial stromal cell ferroptosis promotes angiogenesis in endometriosis. Cell Death Discov. 8:292022. View Article : Google Scholar : PubMed/NCBI | |
|
Wang X, Wei Y, Wei F and Kuang H: Regulatory mechanism and research progress of ferroptosis in obstetrical and gynecological diseases. Front Cell Dev Biol. 11:11469712023. View Article : Google Scholar : PubMed/NCBI | |
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI | |
|
Yadav AK, Yadav PK, Chaudhary GR, Tiwari M, Gupta A, Sharma A, Pandey AN, Pandey AK and Chaube SK: Autophagy in hypoxic ovary. Cell Mol Life Sci. 76:3311–3322. 2019. View Article : Google Scholar : PubMed/NCBI |