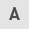‘Reverse Warburg effect’ of cancer‑associated fibroblasts (Review)
- Authors:
- Published online on: April 14, 2022 https://doi.org/10.3892/ijo.2022.5357
- Article Number: 67
Abstract
Introduction
In 1927, Warburg et al proposed the ‘Warburg effect’, which states that even in the case of sufficient oxygen, compared with normal tissues, the glucose metabolism in tumor cells is significantly enhanced (1,2). However, the behavior of some tumor cells is different from the Warburg effect. These tumor cells can oxidize mitochondria to metabolize the oxidative phosphorylation system (OXPHOS) (3), and glycolysis produces ATP at 1 to 64%. OXPHOS remains the main ATP supplier in cancer cells (4). In addition, numerous studies have shown that OXPHOS and aerobic glycolysis are not always mutually exclusive. To some extent, their contribution to ATP production varies with the tumor environment, such as under normoxia and hypoxia (5,6). Therefore, studies have proposed a ‘two-compartment’ model, also known as the ‘Reverse Warburg effect’, to reassess the metabolic pattern of tumor cells (7,8). In this model, cancer cells secrete hydrogen peroxide into the tumor microenvironment (TME) to induce oxidative stress in neighboring stromal cells, so that cancer-associated fibroblasts (CAFs) undergo aerobic glycolysis and produce high levels of energy-rich intermediate metabolites (such as pyruvate and ketones). These energy-rich intermediate metabolites will be used in the mitochondrial TCA cycle and OXPHOS in cancer cells to produce a large quantity of ATP (9,10), such that tumor cells have a higher proliferation ability, which establishes the metabolic interaction between the tumor and the stromal cells. It enables tumor cells to better adapt to changes in oxygen levels, switch metabolic states between glycolysis and oxidative phosphorylation, and survive. Tumor cells exhibit the Warburg effect under hypoxic conditions and the ‘Reverse Warburg effect’ under normoxic conditions, which support tumor growth and metastasis (11).
In addition, in the TME, CAFs are the main tumor stromal cells, which are prone to metabolic reprogramming, and play an important role in tumorigenesis, progression, and metastasis (9,12). Therefore, it is important to study the differentiation of CAFs and the metabolic reprogramming during their crosstalk with cancer cells, which will help to develop new therapeutic strategies and targets (9,13). In the present review, the metabolic reprogramming mechanism of CAFs and cancer crosstalk were summarized, and potential therapeutic strategies involving CAFs reprogramming-associated molecules was also explored.
Literature searches were performed using the PubMed database (https://pubmed.ncbi.nlm.nih.gov/). The key words searched for included: ‘CAFs’, ‘cancer-associated fibroblasts’, ‘metabolic reprogramming’, ‘oxidative stress’, ‘Cav-1’, ‘Warburg effect’, ‘cancer crosstalk’, ‘mitochondrial oxidative phosphorylation’, and ‘reverse Warburg effect’. There were 133 studies covering 1956 to 2022, that were included in the present review.
Origin and differentiation of CAFs
CAFs originate from the activation of resident fibroblasts or other precursor cells. CAFs can be derived from bone marrow mesenchymal stem cells, epithelial cells, cancer cells, endothelial cells, pericytes, smooth muscle cells, adipocytes, fibroblasts, or certain special cells, such as stellate cells, pancreas and liver cells, myoepithelial cells in the breast, and crypt myofibroblasts in the gastrointestinal tract (14). The diversity of sources indicates the heterogeneity of CAFs. Numerous factors can activate CAFs, including cancer cell-derived transforming growth factor-β (TGF-β), epidermal growth factor (EGF), platelet-derived growth factor (PDGFα, PDGFβ), basic fibroblast growth factor (bFGF, also called FGF2), interleukin-6 (IL-6) and interleukin-1β (IL-1β) (15–17), as well as environmental stimuli such as hypoxia, oxidative stress, and the cell matrix. These stimuli may jointly determine different CAF phenotypes, further leading to CAF heterogeneity (18,19). After CAFs are activated, they secrete a large number of growth factors, such as hepatocyte growth factor (HGF), EGF, connective tissue growth factor (CTGF), and insulin-like growth factor (IGF), cytokines, including CXCL12 and IL-6, extracellular vesicles (EVs), metabolites, extracellular matrix (ECM) components, especially collagen, fibronectin, and troponin C (TNC), and ECM remodeling enzymes, such as matrix metalloproteinase (MMP), lysyl oxidase (LOX), and thyroid microsomes (TGMs). These will directly affect the behavior of surrounding cells and reshape the ECM (20). Therefore, CAFs contribute to tumor development, metastasis, tumor metabolism, TME remodeling, and resistance to treatment (21).
CAF differentiation is mainly triggered by factors secreted by cancer cells, but it is also induced by mediators produced by other cell types (such as immune cells). The most important of these is the ‘classical’ TGF-β signaling pathway, which activates the transcription factor Smad, an essential factor for the activation of myofibroblasts. However, the activation of small GTPase RhoA by the ‘non-classical’ signaling pathway also plays a regulatory role. TGF-β depends on the cell type and crosstalk with other signaling pathways, transcription factors, and epigenetic modifications (22). In addition, the differentiation of CAFs may also be affected by released cytokines and growth factors. Another important process of CAF differentiation is metabolic reprogramming, which is related to multiple processes, including oxidative stress, induced by cancer cells. Oxidative stress changes mitochondrial function, leading to higher glucose uptake and levels of reactive oxygen species (ROS), ultimately leading to CAF differentiation (23). However, metabolic reprogramming of tumor-stromal interactions is currently less discussed and rarely recognized as a potential therapeutic avenue. Therefore, both the activation and differentiation of CAFs are inseparable from tumor cells, and are affected by tumor cells and secreted factors, and mediators related to the microenvironment. In turn, CAFs affect tumor cells and form a mutual influence circle.
‘Reverse Warburg effect’ of CAFs
In the 1927, Otto Warburg (2) first proposed the ‘Warburg effect’. Even in the presence of sufficient oxygen, malignant tumor cells tend to produce adenosine triphosphate (ATP). Glycolysis replaces OXPHOS (24), also known as ‘aerobic glycolysis’. The Warburg effect is different, and some tumor cells show a high proportion of OXPHOS (3). In addition, numerous studies have shown that OXPHOS and aerobic glycolysis are not always mutually exclusive. To a certain extent, their contribution to ATP production varies with the tumor environment, such as under normoxia and hypoxia (5,6). The interaction between cancer cells and the surrounding CAFs markedly affects the growth, metabolism, metastasis, and progression of cancer (25). Therefore, a ‘two-compartment’ model, also known as the ‘Reverse Warburg effect’, was proposed to reconsider the metabolism in tumors (7,8). Cancer cells secrete hydrogen peroxide (H2O2) into the TME to induce oxidative stress in neighboring stromal cells. The CAFs derived from stromal cells undergo aerobic glycolysis and produce high levels of energy-rich mitochondrial ‘fuels’ (such as pyruvate, ketone bodies, fatty acids, and lactic acid). In turn, these energy-rich ‘fuels’ are then ‘fed’ to mitochondria in cancer cells, where they are oxidatively metabolized via OXPHOS to produce large quantities of ATP (9,10). Among them, lactic acid is the most important metabolic ‘fuel’. The lactic acid produced by hypoxic cancer cells and CAFs is transported through the lactic acid transporter monocarboxylic acid transporter (MCT)4, then absorbed through MCT1, and distributed to the cancer cells (26,27). After entering cancer cells, lactic acid is metabolized via mitochondrial OXPHOS to produce large quantities of ATP (28), such that cancer cells can achieve self-sufficiency (29). Therefore, the ‘Reverse Warburg effect’ is proposed to link the microenvironment of stromal cells with cancer cells, and provide energy for cancer cells. When there is sufficient oxygen in the tumor microenvironment, tumor cells will experience the ‘Warburg effect’ (30), and pyruvate is converted to lactate by lactate dehydrogenase (LDH) (27,31). The excess lactate produced is taken up by CAF, which increases MCT1 and LDH-1 expression and PDH activity in CAF cells; CAFs incorporate excess lactate into mitochondria for oxidative phosphorylation, thereby removing lactate from the tumor microenvironment (32). When the tumor microenvironment is hypoxic, CAFs undergo metabolic reprogramming and glycolysis, and export lactate to tumor cells for oxidative phosphorylation, providing energy for tumor cells and promoting their proliferation (11).
In the theory of the ‘autophagic tumor stromal model of cancer metabolism’, cancer cells secrete ROS in the TME and induce oxidative stress in CAFs, leading to autophagy and the production of autophagosomes fused with lysosomes. This in turn degrades mitochondria and caveolin 1 (Cav-1). However, the absence of Cav-1 will cause more ROS to be produced in cancer cells, which initiates the oxidative stress cascade in CAFs through a positive feedback mechanism (33). In pancreatic cancer (PDAC), knockdown of Cav-1 promotes ROS production, which in turn reduces Cav-1 expression. Thereby promoting PDAC growth and inducing stroma-tumor metabolic coupling in PDAC (34). Of course, there may be other mechanisms involved in the dysregulation of Cav-1 in CAFs. Studies have found that activation of the TGF-β signaling pathway, the inactivation of tumor suppressor genes (such as p53), and oncogenes (e.g., h-ras, v-abl, brc-abl, and TGF) (35) also participate. Cav-1 binds to nitric oxide synthase (NOS) and inhibits its activity in stromal cells. When Cav-1 is absent, CAFs cannot limit the synthesis of NO. In addition, the accumulation of NO can lead to mitochondrial dysfunction and glycolytic metabolism (36,37). Cav-1 is a negative regulator of ROS produced by the NADPH oxidase (NOX) enzymes, which is achieved through a variety of mechanisms. Cav-1 inhibits NOX2 and NOX5 from producing ROS by directly binding to these enzymes. In addition, Cav-1 can also inhibit NOX2 and NOX4 gene expression and protein synthesis by inhibiting the nuclear factor-κB (NF-κB) pathway (38). The same pathway regulates Cav-1 and ROS, and both contribute to the metabolic transformation of CAFs from mitochondrial OXPHOS to glycolysis (39). In addition, Cav-1 knockdown induces the expression of pyruvate kinase M (PKM)2, which in turn triggers aerobic glycolysis and regulates mitochondrial OXPHOS. The latest study found that Cav-1 directly activates E2F1 in CAFs of lung cancer, which in turn regulates BNIP3, which ultimately induces mitochondrial defects and regulates the metabolic reprogramming pathway of tumor cells (40).
For the metabolic crosstalk between CAFs and cancer cells, hypoxia inducible factor 1α (HIF-1α) and nuclear NF-κB are particularly important. The activation of HIF-1α and NF-κB is mainly mediated by the decreased expression of prolyl hydroxylase domain protein (PHD) (41). Hypoxia has been found to reduce levels of the methyl donor S-adenosylmethionine (42). PKM2 directly interacts with the HIF-1α subunit, promoting the transactivation of HIF-1α target gene by enhancing p300 recruitment and enhancing the binding of HIF-1 to hypoxia response elements. In addition, prolyl hydroxylase 3 (PHD3) interacts with PKM2, which can enhance the combination of PKM2 and HIF-1α, enhance the function of PKM2 coactivator, and hydroxylated PKM2 proline 403/408. Knockdown of PHD3 inhibits PKM2 coactivator function, reduces glucose intake and lactic acid production, and increases O2 consumption in cancer cells. Moreover, JMJD5 interacts directly with PKM2. JMJD5-PKM2 interaction is located in the interface region between PKM2 subunits, blocking PKM2 tetramerization and blocking pyruvate kinase activity. JMJD5-PKM2 interaction makes PKM2 shift to the nucleus and promotes HIF-1α mediated transactivation. JMJD5 and PKM2 are co-recruited to HRE sites of LDHA and PKM2 loci, which promotes HIF-1α-mediated activity and reprograms glucose metabolism in cancer cells (43,44). It was found that oxidative stress triggers the activation of NF-κB and STAT3 in CAFs to upregulate CCL2, thereby promoting oral cancer growth (45). IL-6 secreted by CAFs can promote the growth, migration and invasion of cancer cells in head and neck squamous cell carcinoma (HNSCC) through the integrin αvβ3/NF-κB pathway. And CAF-driven pro-inflammatory signaling is dependent on NF-κB (46). These data suggest that CAF-driven NF-κB signaling plays a central role in mediating inflammation in tumor precursors.
HIF-1α mediates Sirtuin1 (SIRT1) signaling to affect aerobic glycolysis, downregulates oxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) or mitochondrial deacetylase SIRT3, and increases inactive superoxidation and the level of acetylation of superoxide dismutase 2 (SOD2) (47,48). In prostate cancer (PCa), lactate released from CAFs unbalances the NAD+/NADH ratio in PCa cells and increases NAD+ levels, which in turn enhances SIRT1-mediated PGC-1α activation and PCa cell mitochondrial mass and activity (49). In turn, this accumulates ROS and PKM2, and increases the ability to regulate mitochondria. PKM2 is oxidized by excessive ROS, is phosphorylated by activated Src kinase, and then migrates to the nucleus where it recruits HIF-1 and the related embryonic chondrocyte expression gene 1 (DEC1), thereby inhibiting the expression of miR-205, resulting in metabolic reprogramming. Mitochondria OXPHOS improves the survival rate and epithelial-mesenchymal transition (EMT) of cancer cells (50).
In CAFs, glycolysis-related enzymes, such as hexokinase 2 (HK2) and 6-phosphofructokinase liver type (PFKL) are significantly upregulated (51–53). HK2 is a key glycolytic enzyme that is overexpressed in tumors and contributes to the ‘Warburg effect’ (54). During the differentiation of CAFs induced by TGF-β1, the level of the HK2 protein increases. HK2 upregulates p27 protein expression through its downstream metabolite α-KG. In turn, p27 inhibits cyclin-dependent kinase 2 (CDK2) and activates the G1/S checkpoint. This regulatory mechanism links glycolysis with cell cycle control; in fact, the HK2 enzyme regulates both glycolysis and cell cycle checkpoints (55). In addition, there are other glycolytic enzymes that are also closely related, such as the two isoforms PKM1 and PKM2, MCT, and LDHA, LDHB (56–58). The upregulation of PKM1 mediates the production of lactic acid in CAFs. The upregulation of PKM2 promotes the autophagy program in CAFs and stimulates ketone body storage (59,60). A previous in vivo study revealed that overexpression of PKM1 promotes tumor-related inflammation, and overexpression of PKM2 can enhance mitochondrial OXPHOS in cancer cells. High levels of LDHs and MCTs will accelerate the production and transportation of energy-rich ‘fuel’ to cancer cells, thereby promoting their survival, progression, and invasion. In addition, knockdown of encoding isocitrate dehydrogenase [NAD(+)] 3 catalytic subunit α (IDH3α) increased glycolysis and inhibited oxidative phosphorylation in fibroblasts (60).
Another previous study revealed that integrin β2 (ITGB2) is highly expressed in the CAFs of oral squamous cell carcinoma (OSCC). ITGB2 enhances the glycolytic activity of CAFs by regulating the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) pathway. The lactic acid produced by ITGB2 in CAFs is absorbed by tumor cells, metabolized to produce NADH, and then oxidized via mitochondrial OXPHOS to produce ATP. In addition, integrin β 4 (ITGB4) is overexpressed in TNBC cells, and cancer cells provide ITGB4 protein to CAFs through exosomes, after which CAFs induce BNIP3L-dependent mitochondrial autophagy to increase glycolysis levels (61).
The crosstalk between cancer cells and CAFs is closely related to cellular metabolic reprogramming, which contributes to the activation of CAFs, cancer growth, and the progression and escape of cancer treatment. Studying the metabolic reprogramming of CAFs will provide a better understanding of the activation process and the interaction between the stroma and cancer cells, and provide new therapeutic strategies for the development of the original tumor activity of CAFs (Fig. 1).

Effect of ‘Reverse Warburg effect’ on metabolic signaling pathways related to CAFs
Studies have shown that the differentiation of CAFs is necessary for the occurrence and development of cancer, which can occur in the early stages of cancer (12), or before genetic changes in epithelial cells, triggering the malignant transformation of neighboring cells (62). Metabolic reprogramming of CAFs has been detected in breast cancer, lung cancer, PCa, gastric cancer, HNSCC, lymphoma and other solid cancers (63). Factors secreted by tumor cells, as well as cytokines and growth factors, will affect the differentiation of CAFs, including TGF-β, EGF, PDGFα, PDGFβ, bFGF, IL-6 and IL-1β (14,64). Therefore, CAFs are closely related to the signal transmission pathways of tumor cells, and a deeper understanding of the signaling mechanisms between them is required. The following summarizes some related important signaling pathways.
ROS-mediated CAFs and the ‘Reverse Warburg effect’ of cancer cells
Metabolic reprogramming is mainly controlled by oxidative stress and hypoxia. The high levels of ROS produced by cancer cells can induce oxidative stress in CAFs, produce autophagosomes fused with lysosomes, destroy mitochondria, and degrade Cav-1 (65,66). A reduction or mutation of Cav-1 (a marker of autophagy, glycolysis, and oxidative stress) is one of the characteristics of fibroblasts in tumor tissues (67). However, downregulation of Cav-1 in CAFs will lead to higher ROS levels in cancer cells, which will induce oxidative stress in CAFs via a positive feedback loop (68). CAFs and cancer cells maintain their tumor-promoting potential through self-stimulation and cross-communication. In addition, downregulation of Cav-1 in fibroblasts is related to the induction of TGF-β signaling (69). Activation of TGF-β downregulates isocitrate dehydrogenase 1 (IDH1), allowing intracellular accumulation of α-ketoglutarate, leading to downregulation of Cav-1 (70). Downregulation of Cav-1 downregulates prolyl hydroxylase domain-containing protein (PHD) and activity (19), while PHD is involved in the hydroxylation of HIF-1α and IκB. Activation of HIF and NF-κB leads to transcription of glycolytic genes (71). In addition, Cav-1 inhibits NOX2 and NOX5 from producing ROS by directly binding to these enzymes. Furthermore, Cav-1 can also inhibit NOX2 and NOX4 gene expression and protein synthesis by inhibiting the NF-κB pathway (38).
Under normal physiological oxygen concentrations, the α-subunit of HIF is hydroxylated by PHD and then degraded by the E3-ligase and Hippel-Lindau (VHL) protein (72). Accumulated ROS or a hypoxic environment inhibits the production of PHD, reduces the hydroxylation level of HIF-1α, and activates it to stabilize the HIF protein. However, the detailed mechanism of this process is not clear (73). HIF-1α induces hypoxia, promotes the transcription of angiogenic factors (such as VEGF), and mediates autophagy, mitochondrial autophagy, and aerobic glycolysis (74,75), thereby affecting metabolic reprogramming. In addition, SIRT3 (a mitochondrial NAD-dependent deacetylase) in CAFs also affects the stability of ROS and HIF (76). A previous study demonstrated that HIF can regulate and drive the glycolysis of CAFs through SIRT3. In addition, HIF-1 regulates several genes involved in glucose metabolism, such as MCT4 and those encoding glucose transporters (GLUT1 and GLUT3) to increase the uptake of glucose by cells, and affects the output of lactic acid (77). HIF also activates PKM2 transcription, which affects the glucose metabolism of epithelial cells (43). Hypoxia also induces oxidative ATM to promote the glycolytic activity of CAFs by phosphorylating GLUT1 at S490 and increasing the expression of PKM2. In addition, the lactic acid produced by CAFs under hypoxic conditions will act as a metabolic conjugate between CAFs and breast cancer cells, and by activating the TGF-β1/p38 MAPK/MMP2/9 signal axis, it promotes the mitochondrial activity of cancer cells, thereby promoting breast cancer cell invasion (78). Therefore, the oxidative stress induced by ROS production has a significant impact on CAF metabolism, driving CAF aerobic glycolysis, which in turn has a profound impact on the TME and affects the metabolic reprogramming of tumor cells. Put simply, CAFs provide cancer cells with a rich array of compounds and induce antioxidant defenses in cancer cells, thereby allowing cancer cells to proliferate.
Increased ROS levels in the tumor environment also induce the activity of the pro-inflammatory transcription factor NF-κB in fibroblasts, resulting in a CAF-like phenotype (79). The NF-κB target gene cyclooxygenase-2 (COX2) is upregulated in solid tumors and CAFs (80). In addition, activation of NF-κB inhibits the accumulation of ROS by GPX, which in turn causes oxidative stress in CAFs (81).
In general, the signaling pathways activated by ROS activate the metabolic reprogramming of CAFs, which in turn affects the TME, and thus the metabolism of tumor cells (Fig. 2).
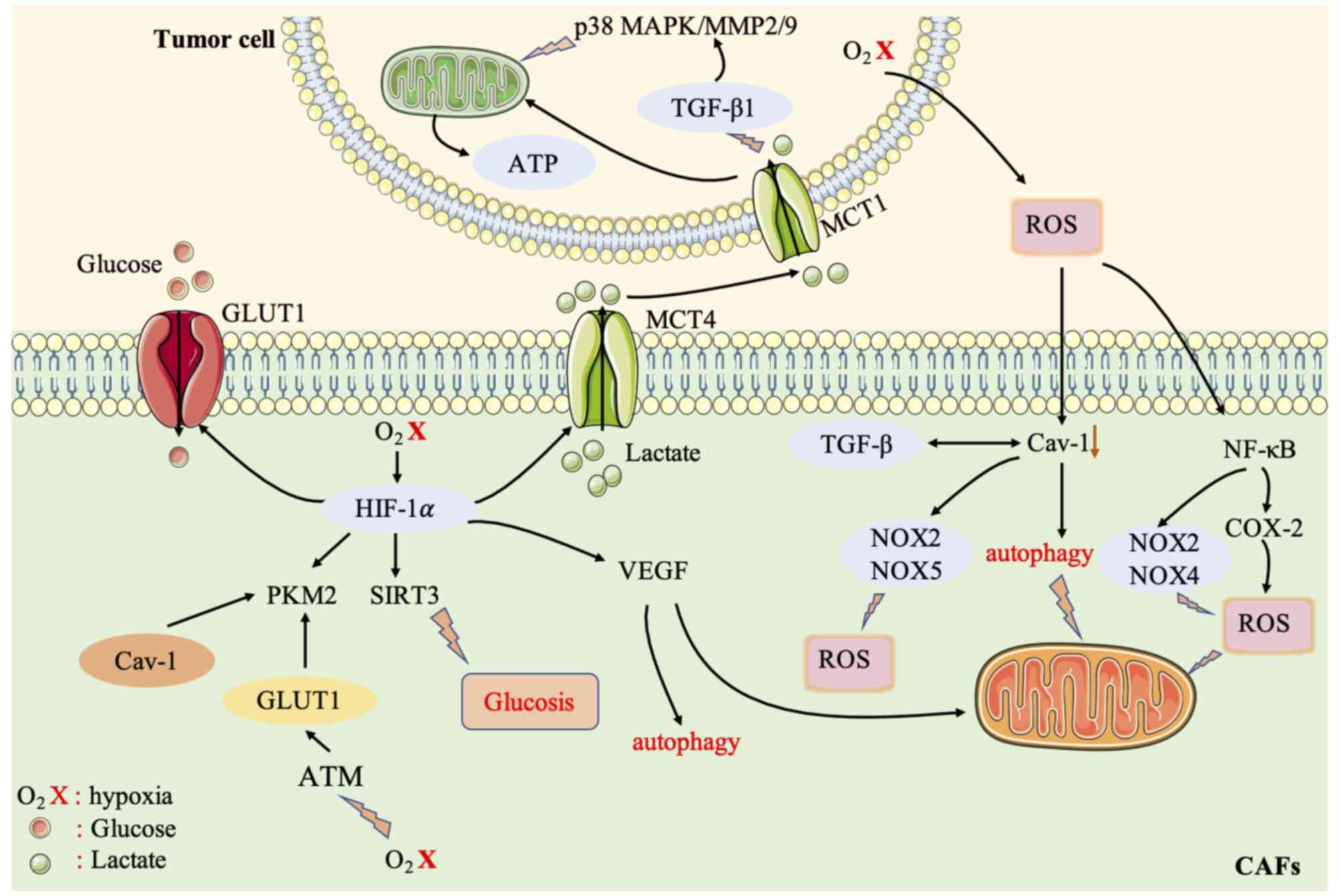
TGF-β signaling pathway mediates the ‘Reverse Warburg effect’ between CAFs and cancer cells
TGF-β plays a key role in the differentiation of CAFs and participates in their differentiation and metabolic regulation (70). The activation of TGF-β in fibroblasts increases oxidative stress, autophagy/mitochondrial autophagy, aerobic glycolysis, and the downregulation of Cav-1, thereby affecting surrounding fibroblasts and supporting cancer cell growth (82). The loss of Cav-1 in stromal cells drives the activation of TGF-β signaling, thereby increasing the transcription of TGF-β target genes, such as encoding connective tissue growth factor (CTGF). Overexpression of CTGF can activate HIF1, promote autophagy, glycolysis, senescence, and metabolism of tumor-associated fibroblasts, thereby promoting tumor growth. On the one hand, TGF-β can regulate the expression of α-SMA in fibroblasts to affect ROS levels (9), stimulate oxidative stress, thus affecting CAFs and tumor cells. On the other hand, TGF-β can upregulate the production of NOX4 and ROS to induce the differentiation of prostate CAFs (83).
The TGF-β signaling pathway is also related to the expression level of some metabolic enzymes, such as IDH1. A study has found that the TGF-β receptor (TGFBR)-IDH1-Cav-1 axis can trigger TGF-β signaling in fibroblasts (56). IDH1 is an enzyme that converts isocitrate into α-ketoglutarate (α-KG) in an NADP+-dependent manner. Downregulation of IDH1 will increase cellular α-KG levels and inhibit Cav-1 expression. Cav-1 downregulation inhibits TGFBR protein degradation and induces TGF-β signaling. Therefore, the TGFBR-IDH1-Cav-1 axis affects fibroblast TGF-β signaling and is an autocrine loop (56).
In addition, some research groups have found that in CAFs, although IDH3α and IDH1 are downregulated, IDH3α knockdown increases glycolysis and inhibits oxidative phosphorylation in fibroblasts, while IDH1 or IDH2 knockdown does not. Moreover, overexpression of IDH3α prevents fibroblasts from converting into CAFs (84). Downregulation of IDH3α reduces the level of α-KG by reducing the ratio of α-KG to fumaric acid and succinic acid, thereby inhibiting PHD2 activity in CAFs, which increases the stability and the level of HIF-1α. The accumulation of HIF-1α in turn increases the uptake of glucose and the production of lactic acid, thus reducing oxygen consumption and promoting glycolysis, and inhibits OXPHOS by upregulating NDUFA4L2, a negative regulator of complex I. This demonstrates that IDH3α is a key metabolic switch in CAFs (84). A recent study revealed that TGF-β type II receptor (TGF-βRII) nuclear translocation of PKM2 inhibits glucose metabolism in CAFs, thereby inhibiting oral cancer tumor growth (85).
A previous study also determined that TGF-β1 downregulates the biosynthesis of acetyl-CoA by regulating the pyruvate dehydrogenase complex (PDC), thereby regulating CAF activation. The reduction of acetyl-CoA and acetylated substrates also leads to the general loss of acetylation of TGF-β1 protein, which in turn affects CAFs (86). It can thus be concluded that the TGF-β signaling pathway is a key metabolic pathway between CAFs and tumor cells, and it is particularly important to understand the relevant mechanisms of the TGF-β signaling pathway in CAFs (Fig. 3).
G protein-coupled estrogen receptor (GPER)-mediated CAFs and the ‘Reverse Warburg effect’ of cancer cells
GPER is a seven-transmembrane-related estrogen receptor belonging to the G protein-coupled receptor family, and is usually upregulated in breast CAFs. GPER, originally known as GPR30, is an alternative estrogen receptor that is structurally distinct from ERα and ERβ and primarily mediates rapid non-genomic responses (87). GPER can regulate cell signaling pathways, and promote breast cancer proliferation, chemoresistance, metastasis, and drug resistance. In breast cancer, 80% of fibroblasts are activated as CAFs (88,89). In vitro experiments have shown that GPER can be expressed in breast cancer CAFs and affects the proliferation and migration of breast cancer cells in response to estrogen signals (90). Therefore, the mechanism of GPER in breast cancer may be related to CAFs in the TME. Compared with normal breast tissues and cells, GPER expression in breast cancer tissues and cells decreased, and GPER expression in CAFs increased. Overexpression of GPER in CAF promotes the expression of Col-1, and its downregulation inhibits the expression of Col-1. The upregulation of GPER in CAFs can promote the proliferation, invasion, and migration of cancer cells, while its downregulation in CAFs has the opposite effects on cancer cells. Therefore, GPER can promote the proliferation, migration, and invasion of triple-negative breast cancer (TNBC) cells through CAFs (91).
Recent research has investigated the role of GPER and hypoxia in the differentiation of CAFs. The expression of GPER in breast CAFs is related to the activation of CAFs induced by hypoxia and the invasion of breast cancer cells. GPER knockdown eliminated hypoxia-driven CAF formation, inhibited breast cancer cell invasion induced by CAF-conditioned medium, and eliminated hypoxia-activated CTGF, VEGF and IL-6 secretion from CAFs (23).
In addition, GPER mediates multidrug resistance in ER positive and negative breast cancer and CAFs in the TME. Thus, it may be a new agent that promotes drug resistance of tumor cells. The PI3K/AKT signaling pathway, activated by breast tumor cells, induces GPER translocation in CAFs in a chromosome region maintenance 1 (CRM1)-dependent manner, and triggers the aerobic glycolysis switch in CAFs through the estrogen/GPER/cAMP/PKA/CREB signal axis. Glycolysis in CAFs supplies additional pyruvate and lactic acid to tumor cells to enhance mitochondrial activity, leading to multi-drug resistance (63).
In summary, GPER in breast CAFs can not only promote the proliferation, migration, and invasion of TNBC cells, but also eliminate the formation of CAFs driven by hypoxia, and affect the crosstalk between CAFs and tumor cells. Notably, the aerobic glycolysis switch in CAFs can be triggered through the estrogen/GPER/cAMP/PKA/CREB signaling axis, such that tumor cells develop drug resistance, suggesting the GPER could be used as a target for drug resistance therapy.
IL-6 signaling molecule mediates the ‘Reverse Warburg effect’ of CAFs and cancer cells
The pro-inflammatory cytokine IL-6 in the TME is closely related to CAFs. IL-6 is a multifunctional cytokine originally considered to be a regulator of immune and inflammatory responses (92). IL-6 exerts its effects by binding to the IL-6α chain and the cytokine receptor signaling subunit gp130, activating the Janus kinase (JAK) family of tyrosine kinases and the signal transducer and activator of transcription (STAT)3 family, thereby regulating target gene transcription (93). Phosphorylation of STAT proteins (1 and 3) can induce the trans-differentiation of normal fibroblasts into CAFs. A previous study showed that when exposed to IL-6 secreted by adjacent gastric cancer cells, primary gastric fibroblasts that constitutively express phosphorylated STAT3 will convert to CAFs (94). In addition, the exosomes exuded by prostate cancer cells under hypoxic conditions contain high levels of IL-6. IL-6 and several other signaling molecules regulate microenvironmental remodeling, EMT, cancer cell stemness, and CAF conversion and differentiation (95). Increased expression of IL-6 has also been reported in CAFs of breast and ovarian tumors (96), indicating that the association between IL-6 and CAFs is particularly important. However, at present, it is not clear how IL-6 regulates the metabolic reprogramming of CAFs. It is worth noting that IL-6 is likely to link inflammation with the enhanced glycolysis in CAFs. This process may be the key to the stimulation of STAT3 by IL-6 and the regulation of certain glycolytic enzymes, such as phosphofructokinase, hexokinase, and fructose-2,6-bisphosphatase (F2,6BP) (97). In addition, studies have found that the transcription factor Twist1 regulates the IL-6 signaling cascade and affects the differentiation of CAFs. When the TME of a solid tumor mass is hypoxic, the overexpression of Twist1 inhibits the senescence of normal fibroblasts and the tumor-promoting effect of CAFs on gastric cancer in the TME. Therefore, Twist1 has been proven to be a highly effective target molecule for anticancer therapy, and the hypoxia-induced Twist1/IL-6 axis provides an important target for regulating the metabolism of CAFs (98,99). Thus, the pro-inflammatory cytokine IL-6 in the TME links inflammation and CAF metabolic reprogramming, which could provide a new target for targeting CAF metabolism, and exhibits favorable development and application prospects.
lncRNAs and microRNAs associated with the ‘Reverse Warburg effect’ of CAFs
Long non-coding RNAs (lncRNAs) are a class of non-coding RNAs with a length of more than 200 nucleotides that have no protein coding ability. LncRNAs participate in various physiological and pathological processes, including development, the immune response, and tumorigenesis (100,101). LncRNAs can control biological processes by interacting with other cellular molecules, including DNA, protein, and RNA. It has been demonstrated that lncRNAs can combine directly with key glycolytic enzymes (102). The enhanced transcription of glycolytic enzyme genes activated by lncRNAs regulates glycolysis in cancer cells (103). In addition, studies have found that the regulation of glycolysis by lncRNAs can be linked to CAFs. CAF-secreted CXCL14 acts on ovarian cancer cells in a paracrine manner. Microarray analysis showed that numerous lncRNAs are dysregulated in ovarian cancer. Among them, lncRNA LINC00092 plays an important role in ovarian cancer cells by interacting with 6-phosphate fructose-2-kinase/fructose-2,6-bisphosphatase 2 (PFKFB2), which increases glycolysis levels and ovarian cancer metastasis. In turn, the glycolytic phenotype of ovarian cancer cells maintains the CAF-like fibroblasts in the TME, forming a regulatory feedback loop in the TME (104). Another lncRNA, urothelial carcinoma associated 1 (UCA1), is induced in glioma cells through the paracrine activity of CXCL14 on glioblastoma stromal cells (105). In addition, the CXCL14-UCA1-miR-182-PFKFB2 axis regulates the interaction between glioma cells and glioblastoma-related stromal cells, and promotes glycolysis and glioma invasion (106).
MicroRNAs (miRNAs) are small noncoding RNAs that regulate numerous biological functions and are key players in regulating cancer development and metastasis. Studies have found that several miRNAs are related to CAF conversion; however, their mechanism of action in CAF cell formation has not yet been fully determined. In addition, some changes in miRNA expression levels have been observed during the formation of activated CAFs in normal fibroblasts (107,108). Among them, the expression levels of miR-221-5p, miR-31-3p, and miR-221-3p were enhanced, and the levels of miR-205, miR-200b, miR200c, miR-141, miR-101, and miR-342 were decreased. These miRNAs affect the conversion of CAFs by regulating the activity of a variety of growth factors, cytokines, and other cell signaling molecules, including HGF, insulin, MAPK, tight and adhesion junction proteins, EGF1, androgen receptor, Wnt, IL-7, and the TGF-β/IL-6 signaling pathway (109).
miRNAs can directly or indirectly regulate the metabolism of glucose and lipids, amino acid biosynthesis, other multi-level and multi-path metabolic enzymes, oncogenes, and tumor suppressor genes, thereby affecting tumor metabolism. Studies have also found that miRNAs can affect the metabolism of tumor stromal cells, thereby affecting tumor progression (110–112). miR-186 is downregulated during the formation of CAFs. This miRNA regulates the level of the membrane-bound glucose transporter GLUT1 by binding to the 3’-untranslated region (UTR) of GLUT1, which promotes its degradation. A study has also found that miR-181c can inhibit glycolysis in CAFs by inhibiting HK2 expression (113). In addition, a study indicated that after miR-21-inhibitor treatment, the degree of glycolysis in CAFs decreased. Following indirect co-culture with CAFs, the oxidative phosphorylation and the expression of SDH, FH, and MCT in BxPc-3 cells increased. After co-culture with miR-21-inhibitor-CAFs, the oxidative phosphorylation and invasion ability of pancreatic cancer cells were reduced. Therefore, miR-21 participates in the metabolic changes of CAFs and affects the development of cancer cells, and thus may be a therapeutic target for the treatment of pancreatic cancer (114).
As relatively recently discovered biomolecules, lncRNAs and miRNAs have attracted increased attention from cancer researchers. They play an important role in the interaction of CAFs with cancer cells, affecting metabolic reprogramming, and can target mRNAs and proteins. As therapeutic targets, they provide a new clinical treatment paradigm.
CAFs and potential therapeutic targets
The crosstalk between cancer cells and CAFs is closely related to the reprogramming of cellular metabolism. By providing the energy needed by tumor cells, CAFs lead to increased mitochondrial activity in tumor cells, and tumor cells acquire drug resistance (23). Therefore, potential therapeutic targets for blocking the metabolic crosstalk between CAFs and tumor cells are very important.
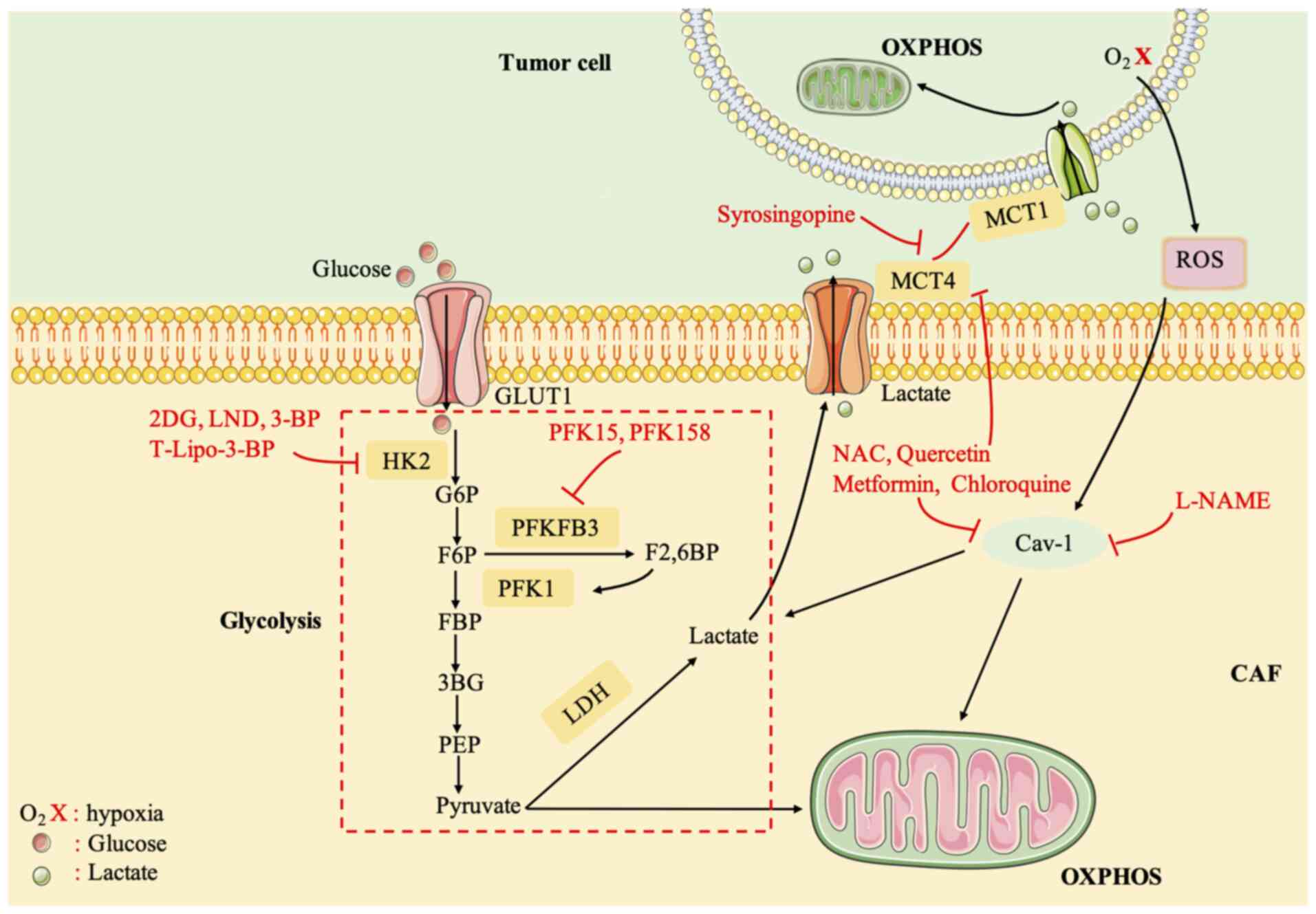
HK2 is an important anticancer drug target (115). Various HK2 inhibitors have been identified, including 2-deoxyglucose (2-DG), 3-bromopyruvate (3-BP), and lonidamine (LND). 2-DG and LND were discontinued because of adverse reactions in clinical trials (116,117). 3-BP can reduce ATP reserves to reverse chemoresistance (118). 3-BP has some adverse reactions, and therefore, it was encapsulated into liposome nanocarriers (T-Lipo-3-BP), which were specifically delivered to the tumor after systemic administration in a mouse tumor model, to eliminate severe side effects and inhibit tumor growth (119). 6-Phosphofructo-2-kinase fructose-2,6-bisphosphatase-3 (PFKFB3) is a potent regulator of glycolysis (120). PFK15, a derivative of a PFKFB3 inhibitor (3PO), was able to induce the rapid induction of apoptosis in transformed cells, exerting an antitumor effect (121). Phase I/II trials of PFK158 in combination with targeted agents are underway (122).
MCT4 is an exporter of glycolysis products and lactate and MCT1 imports lactate into tumors (123). The inhibition of MCT1/MCT4 blocks the metabolic crosstalk between CAFs and tumor cells (124). Syrosingopine, a dual MCT1/MCT4 inhibitor, reduced NAD+ levels by causing lactate accumulation and inhibiting LDH in a mouse model of liver cancer (124). Most other current small molecule MCT inhibitors target MCT1, and one drug, AZD3965, is currently in clinical trials. However, the drug was revealed to be ineffective in the presence of MCT4 expression (125). A clinical trial demonstrated that the antioxidant N-acetylcysteine (NAC) can reduce the expression of MCT4 in CAFs of patients with cancer and reduces the proliferation rate of cancer cells in women with stage 0 and stage I breast cancer. It is effective and safe in breast cancer treatment (126). In addition, quercetin, metformin, and chloroquine also significantly reduced the expression of MCT4 in CAFs (19,127,128). In some patients with aggressive B-cell lymphoma with lactic acidosis, metformin and sirolimus can inhibit the activation of the ‘lactate shuttle’ (or high expression of MCT4), reduce the expression of LDHB and PKM1, and inhibit CAF-carcinoma cellular metabolism coupling (129).
Antioxidant treatment can also block the metabolic cross-talk between CAFs and tumor cells, which should inhibit ROS (122). NAC treatment effectively inhibits tumor growth by preventing DNA damage and genetic instability, and reducing HIF-1 levels (130). Metformin acts as a complex I inhibitor and blocks mitochondria-dependent ROS production (131). In addition, NAC, metformin, L-NAME (a nitric oxide NO inhibitor), quercetin, and chloroquine can restore Cav-1 expression, thereby inhibiting ROS generation. Clinical trials of metformin alone and in combination with standard therapy are ongoing in multiple tumor types (19,132). Furthermore, based on the regulation of Cav-1, MCT4, and HIF-1α expression in CAFs, the combination of acetylcysteine (N-acetyl-L-cysteine) and topotecan is being tested in patients with ovarian cancer in phase II clinical trials (Fig. 4) (133).
Conclusions
The occurrence and development of tumors are related to the metabolic reprogramming of the TME, in which the metabolic reprogramming of CAFs is very important. The metabolic reprogramming of CAFs activates a variety of signaling pathways that act on tumor cells, and in turn, tumor cells will also affect CAFs, such as in the differentiation and the activation of related signaling pathways. The metabolic reprogramming of CAFs is closely related to the proliferation, metastasis, angiogenesis, drug resistance, and other aggressive behaviors of cancer cells. Therefore, the metabolic reprogramming of CAFs and cancer crosstalk increase the heterogeneity and plasticity of cancer metabolism. This will closely link the study of tumor cells with the TME, and help researchers to study the occurrence and development of tumors, providing a series of new predictive biomarkers and strategies for anticancer treatment. Therefore, in-depth study of the metabolic reprogramming of CAFs is of great significance and will provide new methods for future cancer treatments.
Acknowledgements
Not applicable.
Funding
This work was supported by the Hunan Provincial Natural Science Foundation (grant no. 2021JJ30915), and the Fundamental Research Funds for the Central Universities of Central South University.
Availability of data and materials
Not applicable.
Authors' contributions
LL wrote the manuscript and drew the figures. XL, XJ, WL, and QL collected the related studies and helped to revise the manuscript. YZ and YL designed and revised the manuscript. Data authentication is not applicable to the present review. All the authors read and approved the final version of the review.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Warburg O: On the origin of cancer cells. Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI | |
|
Warburg O, Wind F and Negelein E: The metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927. View Article : Google Scholar : PubMed/NCBI | |
|
Jiang P, Du W and Wu M: Regulation of the pentose phosphate pathway in cancer. Protein Cell. 5:592–602. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Zheng J: Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett. 4:1151–1157. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Alfarouk KO, Shayoub ME, Muddathir AK, Elhassan GO and Bashir AH: Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (Dismantling of Multicellularity). Cancers (Basel). 3:3002–3017. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Xu XD, Shao SX, Jiang HP, Cao YW, Wang YH, Yang XC, Wang YL, Wang XS and Niu HT: Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol Res Treat. 38:117–122. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Yoshida GJ: Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 34:1112015. View Article : Google Scholar : PubMed/NCBI | |
|
Saada A: Mitochondria: Mitochondrial OXPHOS (dys) function ex vivo-the use of primary fibroblasts. Int J Biochem Cell Biol. 48:60–65. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Arcucci A, Ruocco MR, Granato G, Sacco AM and Montagnani S: Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. Biomed Res Int. 2016:45028462016. View Article : Google Scholar : PubMed/NCBI | |
|
Pertega-Gomes N, Vizcaino JR, Attig J, Jurmeister S, Lopes C and Baltazar F: A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer. 14:3522014. View Article : Google Scholar : PubMed/NCBI | |
|
Lee M and Yoon JH: Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J Biol Chem. 6:148–161. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Buchsbaum RJ and Oh SY: Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers (Basel). 8:192016. View Article : Google Scholar : PubMed/NCBI | |
|
Catalano V, Turdo A, Di Franco S, Dieli F, Todaro M and Stassi G: Tumor and its microenvironment: A synergistic interplay. Semin Cancer Biol. 23:522–532. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Santi A, Kugeratski FG and Zanivan S: Cancer associated fibroblasts: The architects of stroma remodeling. Proteomics. 18:e17001672018. View Article : Google Scholar : PubMed/NCBI | |
|
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L and Chiarugi P: Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 70:6945–6956. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Lohr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H and Jesnowski R: Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 61:550–555. 2001.PubMed/NCBI | |
|
Shao ZM, Nguyen M and Barsky SH: Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 19:4337–4345. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G and Sahai E: Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 15:637–646. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al: Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 9:3256–3276. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Cirri P and Chiarugi P: Cancer associated fibroblasts: The dark side of the coin. Am J Cancer Res. 1:482–497. 2011.PubMed/NCBI | |
|
Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R and Sahai E: Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 27:574–588. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Massague J: TGFβ signalling in context. Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Avagliano A, Granato G, Ruocco MR, Romano V, Belviso I, Carfora A, Montagnani S and Arcucci A: Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. Biomed Res Int. 2018:60754032018. View Article : Google Scholar : PubMed/NCBI | |
|
Marin D and Sabater B: The cancer Warburg effect may be a testable example of the minimum entropy production rate principle. Phys Biol. 14:0240012017. View Article : Google Scholar : PubMed/NCBI | |
|
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL and Weinberg RA: Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Pereira-Nunes A, Afonso J, Granja S and Baltazar F: Lactate and lactate transporters as key players in the maintenance of the warburg effect. Adv Exp Med Biol. 1219:51–74. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Draoui N and Feron O: Lactate shuttles at a glance: From physiological paradigms to anti-cancer treatments. Dis Model Mech. 4:727–732. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Lee N and Kim D: Cancer metabolism: Fueling more than just growth. Mol Cells. 39:847–854. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Wilson RB, Solass W, Archid R, Weinreich FJ, Konigsrainer A and Reymond MA: Resistance to anoikis in transcoelomic shedding: The role of glycolytic enzymes. Pleura Peritoneum. 4:201900032019. View Article : Google Scholar : PubMed/NCBI | |
|
Hsu PP and Sabatini DM: Cancer cell metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Konjević G, Jurisić V, Jakovljević B and Spuzić I: Lactate dehydrogenase (LDH) in peripheral blood lymphocytes (PBL) of patients with solid tumors. Glas Srp Akad Nauka Med. 137–147. 2002.(In Serbian). PubMed/NCBI | |
|
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC and Harris AL: Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 7:1–6. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Chen D and Che G: Value of caveolin-1 in cancer progression and prognosis: Emphasis on cancer-associated fibroblasts, human cancer cells and mechanism of caveolin-1 expression (Review). Oncol Lett. 8:1409–1421. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Shao S, Qin T, Qian W, Yue Y, Xiao Y, Li X, Zhang D, Wang Z, Ma Q and Lei J: Positive feedback in Cav-1-ROS signalling in PSCs mediates metabolic coupling between PSCs and tumour cells. J Cell Mol Med. 24:9397–9408. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Bist A, Fielding CJ and Fielding PE: p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry. 39:1966–1972. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, Pestell RG, Sotgia F, Rui H and Lisanti MP: Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther. 8:1071–1079. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, et al: The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 9:3485–3505. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Chen F, Barman S, Yu Y, Haigh S, Wang Y, Black SM, Rafikov R, Dou H, Bagi Z, Han W, et al: Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. 73:201–213. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG and Lisanti MP: Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 13:2132011. View Article : Google Scholar : PubMed/NCBI | |
|
Shen C, Chen X, Xiao K and Che G: New relationship of E2F1 and BNIP3 with caveolin-1 in lung cancer-associated fibroblasts. Thorac Cancer. 11:1369–1371. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Semenza GL: HIF-1: Upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Q, Liu L, Zhao Y, Zhang J, Wang D, Chen J, He Y, Wu J, Zhang Z, Liu Z, et al: Hypoxia induces genomic DNA demethylation through the activation of HIF-1α and transcriptional upregulation of MAT2A in hepatoma cells. Mol Cancer Ther. 10:1113–1123. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ and Wang WC: JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism. Proc Natl Acad Sci USA. 111:279–284. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Li X, Xu Q, Wu Y, Li J, Tang D, Han L and Fan Q: A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis. 35:1362–1370. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Qin X, Yan M, Wang X, Xu Q, Wang X, Zhu X, Shi J, Li Z, Zhang J, Chen W, et al: Cancer-associated Fibroblast-derived IL-6 promotes head and neck cancer progression via the osteopontin-NF-kappa B signaling pathway. Theranostics. 8:921–940. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Chiarugi P and Cirri P: Metabolic exchanges within tumor microenvironment. Cancer Lett. 380:272–280. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P and Chiarugi P: Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 72:5130–5140. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A, Raspollini MR, Magherini F, Rapizzi E, Masquelier J, et al: Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene. 38:5339–5355. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez-Outschoorn U, Sotgia F and Lisanti MP: Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Semin Oncol. 41:195–216. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, et al: The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 8:3984–4001. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Roy A and Bera S: CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumour Biol. 37:8503–8514. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Hasebe T, Mukai K, Tsuda H and Ochiai A: New prognostic histological parameter of invasive ductal carcinoma of the breast: Clinicopathological significance of fibrotic focus. Pathol Int. 50:263–272. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Mao Y, Keller ET, Garfield DH, Shen K and Wang J: Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 32:303–315. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Hu JW, Sun P, Zhang DX, Xiong WJ and Mi J: Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal. 26:2210–2216. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, et al: Transcriptional evidence for the ‘Reverse Warburg Effect’ in human breast cancer tumor stroma and metastasis: Similarities with oxidative stress, inflammation, Alzheimer's disease, and ‘Neuron-Glia Metabolic Coupling’. Aging (Albany NY). 2:185–199. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe R, Howell A, Pestell RG, Smith J, Daniel R, Sotgia F and Lisanti MP: Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther. 12:1101–1113. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D and Chiarugi P: Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget. 6:24061–24074. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Li Z, Yang P and Li Z: The multifaceted regulation and functions of PKM2 in tumor progression. Biochim Biophys Acta. 1846:285–296. 2014.PubMed/NCBI | |
|
Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, et al: Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci USA. 111:15526–15531. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG and Cho NH: ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene. 39:664–676. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Qiao A, Gu F, Guo X, Zhang X and Fu L: Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front Med. 10:33–40. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA, Guo L, Zhu Q, Luo H, Du YE, et al: Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 36:2131–2145. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Ziani L, Chouaib S and Thiery J: Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. 9:4142018. View Article : Google Scholar : PubMed/NCBI | |
|
Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, Li G and Zhou M: The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. 8:57813–57825. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez-Outschoorn UE, Lisanti MP and Sotgia F: Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 25:47–60. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Dabiri S, Talebi A, Shahryari J, Meymandi MS and Safizadeh H: Distribution of myofibroblast cells and microvessels around invasive ductal carcinoma of the breast and comparing with the adjacent range of their normal-to-DCIS zones. Arch Iran Med. 16:93–99. 2013.PubMed/NCBI | |
|
Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, Sneddon S, Pestell RG, Martinez-Outschoorn U, Lisanti MP and Sotgia F: CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 11:2272–2284. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Khan HY and Orimo A: Transforming growth factor-β: Guardian of catabolic metabolism in carcinoma-associated fibroblasts. Cell Cycle. 11:4302–4303. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Hou X, Zhang J, Wang Y, Xiong W and Mi J: TGFBR-IDH1-Cav1 axis promotes TGF-β signalling in cancer-associated fibroblast. Oncotarget. 8:83962–83974. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Trimmer C, Sotgia F, Whitaker-Menezes D, Balliet RM, Eaton G, Martinez-Outschoorn UE, Pavlides S, Howell A, Iozzo RV, Pestell RG, et al: Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: A new genetically tractable model for human cancer associated fibroblasts. Cancer Biol Ther. 11:383–394. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Panday A, Inda ME, Bagam P, Sahoo MK, Osorio D and Batra S: Transcription factor NF-κB: An update on intervention strategies. Arch Immunol Ther Exp (Warsz). 64:463–483. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Zwaans BM and Lombard DB: Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Model Mech. 7:1023–1032. 2014.PubMed/NCBI | |
|
De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A and Maggiolini M: HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 15:R642013. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou F, Du J and Wang J: Albendazole inhibits HIF-1α-dependent glycolysis and VEGF expression in non-small cell lung cancer cells. Mol Cell Biochem. 428:171–178. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Fiaschi T and Chiarugi P: Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int J Cell Biol. 2012:7628252012. View Article : Google Scholar : PubMed/NCBI | |
|
Ullah MS, Davies AJ and Halestrap AP: The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 281:9030–9037. 2006. View Article : Google Scholar : PubMed/NCBI | |
|
Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, Peng M, Zhao M, Cui X and Liu M: Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 41:370–383. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Erez N, Truitt M, Olson P, Arron ST and Hanahan D: Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 17:135–147. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Zhu Y, Shi C, Zeng L, Liu G, Jiang W, Zhang X, Chen S, Guo J, Jian X, Ouyang J, et al: High COX-2 expression in cancer-associated fibiroblasts contributes to poor survival and promotes migration and invasiveness in nasopharyngeal carcinoma. Mol Carcinog. 59:265–280. 2020. View Article : Google Scholar : PubMed/NCBI | |
|
Chan JS, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC, Li L, Zhu P and Tan NS: Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 8:e25622017. View Article : Google Scholar : PubMed/NCBI | |
|
Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S, Martinez-Outschoorn U, et al: Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with ‘Warburg-like’ cancer metabolism and L-lactate production. Cell Cycle. 11:3019–3035. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Sampson N, Koziel R, Zenzmaier C, Bubendorf L, Plas E, Jansen-Dürr P and Berger P: ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol Endocrinol. 25:503–515. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, Zhang J, Zhao S, Zhou BP and Mi J: Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 10:1335–1348. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Wu F, Wang S, Zeng Q, Liu J, Yang J, Mu J, Xu H, Wu L, Gao Q, He X, et al: TGF-βRII regulates glucose metabolism in oral cancer-associated fibroblasts via promoting PKM2 nuclear translocation. Cell Death Discov. 8:32022. View Article : Google Scholar : PubMed/NCBI | |
|
Smith ER and Hewitson TD: TGF-β1 is a regulator of the pyruvate dehydrogenase complex in fibroblasts. Sci Rep. 10:179142020. View Article : Google Scholar : PubMed/NCBI | |
|
Pupo M, Maggiolini M and Musti AM: GPER mediates non-genomic effects of estrogen. Methods Mol Biol. 1366:471–488. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Madeo A and Maggiolini M: Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 70:6036–6046. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Vivacqua A, Romeo E, De Marco P, De Francesco EM, Abonante S and Maggiolini M: GPER mediates the Egr-1 expression induced by 17β-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res Treat. 133:1025–1035. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
De Francesco EM, Sims AH, Maggiolini M, Sotgia F, Lisanti MP and Clarke RB: GPER mediates the angiocrine actions induced by IGF1 through the HIF-1alpha/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res. 19:1292017. View Article : Google Scholar : PubMed/NCBI | |
|
Yang K and Yao Y: Mechanism of GPER promoting proliferation, migration and invasion of triple-negative breast cancer cells through CAF. Am J Transl Res. 11:5858–5868. 2019.PubMed/NCBI | |
|
Grivennikov SI, Greten FR and Karin M: Immunity, inflammation, and cancer. Cell. 140:883–899. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Bromberg J and Wang TC: Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Kinoshita H, Hirata Y, Nakagawa H, Sakamoto K, Hayakawa Y, Takahashi R, Nakata W, Sakitani K, Serizawa T, Hikiba Y, et al: Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS One. 8:e609142013. View Article : Google Scholar : PubMed/NCBI | |
|
Ramteke A, Ting H, Agarwal C, Mateen S, Somasagara R, Hussain A, Graner M, Frederick B, Agarwal R and Deep G: Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol Carcinog. 54:554–565. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Erez N, Glanz S, Raz Y, Avivi C and Barshack I: Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 437:397–402. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Ando M, Uehara I, Kogure K, Asano Y, Nakajima W, Abe Y, Kawauchi K and Tanaka N: Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J Nippon Med Sch. 77:97–105. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Khan MA, Chen HC, Zhang D and Fu J: Twist: A molecular target in cancer therapeutics. Tumour Biol. 34:2497–2506. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Lee KW, Yeo SY, Sung CO and Kim SH: Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Res. 75:73–85. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Schmitz SU, Grote P and Herrmann BG: Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 73:2491–2509. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Garen A: From a retrovirus infection of mice to a long noncoding RNA that induces proto-oncogene transcription and oncogenesis via an epigenetic transcription switch. Signal Transduct Target Ther. 1:160072016. View Article : Google Scholar : PubMed/NCBI | |
|
Ma MZ, Zhang Y, Weng MZ, Wang SH, Hu Y, Hou ZY, Qin YY, Gong W, Zhang YJ, Kong X, et al: Long Noncoding RNA GCASPC, a Target of miR-17-3p, negatively regulates pyruvate carboxylase-dependent cell proliferation in gallbladder cancer. Cancer Res. 76:5361–5371. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Hanahan D and Weinberg RA: Hallmarks of cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Zhao L, Ji G, Le X, Wang C, Xu L, Feng M, Zhang Y, Yang H, Xuan Y, Yang Y, et al: Long Noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 77:1369–1382. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
He Z, You C and Zhao D: Long non-coding RNA UCA1/miR-182/PFKFB2 axis modulates glioblastoma-associated stromal cells-mediated glycolysis and invasion of glioma cells. Biochem Biophys Res Commun. 500:569–576. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Ahn YH and Kim JS: Long Non-Coding RNAs as regulators of interactions between cancer-associated fibroblasts and cancer cells in the tumor microenvironment. Int J Mol Sci. 21:74842020. View Article : Google Scholar : PubMed/NCBI | |
|
Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME and Lengyel E: MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2:1100–1108. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, Yu BQ, Zhi QM, Li CL, Wang YQ, et al: Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 25:588–603. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Zeng Z, Hu P, Tang X, Zhang H, Du Y, Wen S and Liu M: Dectection and analysis of miRNA expression in breast cancer-associated fibroblasts. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 30:1071–1075. 2014.(In Chinese). PubMed/NCBI | |
|
Wang Z, Tan Y, Yu W, Zheng S, Zhang S, Sun L and Ding K: Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int J Biol Sci. 13:339–348. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Vallabhaneni KC, Hassler MY, Abraham A, Whitt J, Mo YY, Atfi A and Pochampally R: Mesenchymal Stem/Stromal cells under stress increase osteosarcoma migration and apoptosis resistance via extracellular vesicle mediated communication. PLoS One. 11:e01660272016. View Article : Google Scholar : PubMed/NCBI | |
|
Tang H, Lee M, Sharpe O, Salamone L, Noonan EJ, Hoang CD, Levine S, Robinson WH and Shrager JB: Oxidative stress-responsive microRNA-320 regulates glycolysis in diverse biological systems. FASEB J. 26:4710–4721. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Sun P, Hu JW, Xiong WJ and Mi J: miR-186 regulates glycolysis through Glut1 during the formation of cancer-associated fibroblasts. Asian Pac J Cancer Prev. 15:4245–4250. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Chen S, Chen X, Shan T, Ma J, Lin W, Li W and Kang Y: MiR-21-mediated Metabolic alteration of cancer-associated fibroblasts and its effect on pancreatic cancer cell behavior. Int J Biol Sci. 14:100–110. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Grasso C, Jansen G and Giovannetti E: Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit Rev Oncol Hematol. 114:139–152. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Kurtoglu M, Maher JC and Lampidis TJ: Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxid Redox Signal. 9:1383–1390. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Nancolas B, Guo L, Zhou R, Nath K, Nelson DS, Leeper DB, Blair IA, Glickson JD and Halestrap AP: The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem J. 473:929–936. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Gatenby RA and Gillies RJ: Glycolysis in cancer: A potential target for therapy. Int J Biochem Cell Biol. 39:1358–1366. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Zhang Y, Wei J, Xu J, Leong WS, Liu G, Ji T, Cheng Z, Wang J, Lang J, Zhao Y, et al: Suppression of tumor energy supply by liposomal nanoparticle-mediated inhibition of aerobic glycolysis. ACS Appl Mater Interfaces. 10:2347–2353. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Lu L, Chen Y and Zhu Y: The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget. 8:62793–62802. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA II, Klarer AC, Redman R, Miller DM, Trent JO, et al: Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 12:1461–1470. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Lisanti MP, Martinez-Outschoorn UE and Sotgia F: Oncogenes induce the cancer-associated fibroblast phenotype: Metabolic symbiosis and ‘fibroblast addiction’ are new therapeutic targets for drug discovery. Cell Cycle. 12:2723–2732. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Lamb R, Ozsvari B, Bonuccelli G, Smith DL, Pestell RG, Martinez-Outschoorn UE, Clarke RB, Sotgia F and Lisanti MP: Dissecting tumor metabolic heterogeneity: Telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget. 6:21892–21905. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, Maira SM, Moroni C, Lane HA and Hall MN: Dual Inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 25:3047–3058.e4. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, Trapani F, Bishop PW, White A, Critchlow SE, et al: Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 20:926–937. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Monti D, Sotgia F, Whitaker-Menezes D, Tuluc M, Birbe R, Berger A, Lazar M, Cotzia P, Draganova-Tacheva R, Lin Z, et al: Pilot study demonstrating metabolic and anti-proliferative effects of in vivo anti-oxidant supplementation with N-Acetylcysteine in Breast Cancer. Semin Oncol. 44:226–232. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Crawford S: Anti-inflammatory/antioxidant use in long-term maintenance cancer therapy: A new therapeutic approach to disease progression and recurrence. Ther Adv Med Oncol. 6:52–68. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, Migneco G, Witkiewicz AK, Martinez-Cantarin MP, Flomenberg N, et al: Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 9:2423–2433. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Martinez-Outschoorn UE, Whitaker-Menezes D, Valsecchi M, Martinez-Cantarin MP, Dulau-Florea A, Gong J, Howell A, Flomenberg N, Pestell RG, Wagner J, et al: Reverse Warburg effect in a patient with aggressive B-cell lymphoma: Is lactic acidosis a paraneoplastic syndrome? Semin Oncol. 40:403–418. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, et al: HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 12:230–238. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Morales AI, Detaille D, Prieto M, Puente A, Briones E, Arévalo M, Leverve X, López-Novoa JM and El-Mir MY: Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int. 77:861–869. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Sonnenblick A, Agbor-Tarh D, Bradbury I, Di Cosimo S, Azim HA Jr, Fumagalli D, Sarp S, Wolff AC, Andersson M, Kroep J, et al: Impact of diabetes, insulin, and metformin use on the outcome of patients with human epidermal growth factor receptor 2-positive primary breast cancer: Analysis from the ALTTO PHASE III randomized Trial. J Clin Oncol. 35:1421–1429. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Cirri P and Chiarugi P: Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 31:195–208. 2012. View Article : Google Scholar : PubMed/NCBI |