Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway
- Authors:
- Published online on: August 13, 2013 https://doi.org/10.3892/mmr.2013.1628
- Pages: 1011-1016
Abstract
Introduction
Hypoxic-ischemic (HI) brain injury is a serious insult that results in various degrees of damage to the body, leading to significant neurobehavioral dysfunctions. The immature brain is particularly fragile to oxygen deprivation (OD), termed HI brain damage (HIBD). Following HI stimulation, whether neurons undergo apoptosis or survive is dependent upon the duration that cells are able to maintain homeostasis following the insult. Autophagy is a cell-autonomous survival mechanism that maintains cell homeostasis. It is an essential catabolic process that degrades misfolded proteins and clears excess or damaged organelles in cells by isolating them in double membrane structures, which are then fused with lysosomes. This process contributes to the maintenance of the balance between degradation, synthesis and the recycling of cellular components, which is essential for cell homeostasis (1). An increasing number of studies have indicated that the induction of cellular autophagy was associated with the attenuation of ischemia/reperfusion (I/R) injury (2,3).
Brain-derived neurotrophic factor (BDNF), a member of the mammalian neurotrophin family, has been demonstrated to be a potent growth factor that is beneficial to neuronal function following HI. The effects of BDNF are mediated via binding to its specific receptors, tyrosine kinase B (TrkB) and p75 (4,5). Mature BDNF triggers three intracellular signaling cascades, MAPK, PI3K and PLCγ pathways, which are its predominant downstream regulators (6). Our previous studies also demonstrated that BDNF prevented cortical neurons from hypoxia-induced neurotoxicity by the upregulation of TrkB mRNA (7) and through the activation of the extracellular signal-regulated kinase (ERK) and PI3K pathways; however, not through activation of the p38 MAPK pathway (8). Akt is involved in multiple signaling pathways that relate to numerous biological processes, including cell proliferation and differentiation. It has been demonstrated that Akt is a central regulator of injury progression due to its extensive signaling in the ischemic brain. The best-studied downstream substrate of Akt is the mammalian target of rapamycin (mTOR), which is significant in inducing mammalian autophagy. Ribosomal protein S6 kinase (p70S6K) is the downstream effector of mTOR and controls protein translation. A study has suggested that the activation of the PI3K-Akt-mTOR signaling pathway may promote necrotic cell death via the suppression of autophagy (9). Furthermore, caffeine induced apoptosis by enhancing autophagy via PI3K/Akt/mTOR/p70S6K inhibition (10) and ophiopogonin B-induced autophagy in non-small cell lung cancer cells via the inhibition of the PI3K/Akt signaling pathway (11).
Based on these previous results, it was hypothesized that BDNF may protect neurons from hypoxia injury in vitro by the induction of autophagy through the PI3K/Akt/mTOR/p70S6K signaling pathway. To test this hypothesis, cortical neurons were isolated from pregnant rats (gestational days 16–18) and cultured. Autophagy following BDNF treatment was investigated by microtubule-associated protein light chain 3 (LC3) conversion and autophagosome formation by LC3 fluorescence. The phosphorylation of Akt, mTOR and p70S6K was detected in cultured cells with or without BDNF treatment. Cell variability was analyzed by the Cell Counting Kit-8 for determination of the protective effect of BDNF.
Materials and methods
Primary culture of cortical neurons
All animal procedures were approved by the Sichuan University Committee on Animal Research (Sichuan, China). Pregnant Sprague-Dawley rats were supplied by the Experimental Animal Center of Sichuan University (Sichuan, China). Each test was repeated 3 times and neurons were cultured from three independent pregnant rats.
The method was used as described previously (12) with modifications. Briefly, cortical neuron cultures were prepared from the brains of rat embryos (gestational days 16–18). Cerebral hemispheres were isolated aseptically and the meninges, olfactory bulbs and hippocampi were removed. The hemispheres were then incubated in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) containing 0.05% trypsin and 10 mg/ml DNase (Roche Diagnostics, Manheim, Germany) at 37°C for 0.5–1 min. The cell suspensions were filtered through a 70 mm Falcon nylon cell strainer (BD Biosciences, Franklin Lakes, NJ, USA) to remove debris and centrifuged for 5 min at 120 × g with a Beckman-Coulter centrifuge (Miami, FL, USA). Cells were resuspended in Neurobasal medium containing 2% B27, 1% Glutamax, 100 IU/ml penicillin and 100 mg/ml streptomycin (Invitrogen Life Technologies, Carlsbad, CA, USA). Cells were then seeded at 1.6×106/ml on 25 ml Corning culture dishes and precoated with 100 mg/ml of poly-D-lysine acidic protein (Sigma-Aldrich) for astrocytes.
Neurons were incubated in a humidified atmosphere of 95% air and 5% CO2 and maintained at 37°C as described previously (8). Half of the medium was then replaced with fresh Neurobasal medium every 3 days. Experiments were initiated in vitro on days 7–10 when neurons were mature [i.e., >95% cells were neurons as determined by immunofluorescence staining with MAP2 for neurons and glial fibrillary acidic protein (GFAP) for astrocytes].
Hypoxia
OD was performed to initiate neonatal HI conditions in order to investigate the induction of autophagy in vitro, since consistent oxygen-glucose deprivation (OGD) destroyed the cells within 2 h in the pre-test. For OD, oxygen was removed by placing the cells in an anaerobic chamber perfused with 99% N2 at 37°C.
Cell viability
Cell viability was assessed by the Cell Counting Kit-8 (CCK-8, Dojindo, Kunamoto, Japan). Neurons dispensed as 100 μl cell suspension were seeded in 96-well plates (50,000 cells/well). The various groups were treated as follows: Group A (control), cultured under hypoxic conditions; Group B (50 ng/ml BDNF) received 50 ng/ml BDNF 30 min before hypoxia; Group C (100 ng/ml BDNF) received 100 ng/ml BDNF 30 min before hypoxia; and Group D (200 ng/ml BDNF) received BDNF 200 ng/ml 30 min before hypoxia. Subsequent to the different treatments, 10 μl CCK-8 solution was added to each well, according to the manufacturer’s instructions. Subsequent to incubation at 37°C for 3 h in a humidified CO2 incubator, absorbance at 450 nm was measured with a microplate reader (Bio-Rad, Hercules, CA, USA). The values were used to calculate cell viability by setting the normoxic control to 100% (13).
Western blot analysis
Total cell extracts were prepared. Protein concentration was determined using the bicinchoninic acid protein assay (Pierce Biotechnology, Inc., Rockford, IL, USA). The protein aliquot (40 μg) was subjected to sodium dodecyl sulfate gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). Subsequent to blocking with 5% non-fat milk, the membranes were incubated with the following primary antibodies: anti-phospho-Akt (Ser473; dilution, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA, #9271), anti-phospho-mTOR (Ser2448; dilution, 1:1,000; Cell Signaling Technology, Inc., #2971), anti-phospho-p70S6K (THR389; dilution, 1:1,000; Cell Signaling Technology, Inc., #9205), anti-β-actin (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and anti-LC3 (dilution, 1:1,000; Cell Signaling Technology, Inc., #4108). The membranes were washed and incubated with horseradish peroxidase-conjugated anti-mouse (dilution, 1:1,000; Bio-Rad; #170-6520) or anti-rabbit secondary antibodies (dilution, 1:1000; Bio-Rad; #166-2408) for 2 h. The membrane was washed and specific bands were detected with Immobilon Western Chemiluminescent horseradish peroxidase substrate (Millipore) and bands were quantified using the Gel-Pro image analyzer software 4.0 (Media Cybernetics, Rockville, MD, USA).
Fluorescence microscopy analysis
The mouse MAP2 antibody (dilution, 1:250; Abcam, Cambridge, MA, USA) and the rabbit LC3 antibody (dilution, 1:500; Abcam) were used for immunocytochemistry 3 h following OD and BDNF exposure. The immunocytochemistry procedure was described previously (6). Images were observed using a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Statistical analysis
Data are presented as the mean ± SD. One-way analysis of variance was performed for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
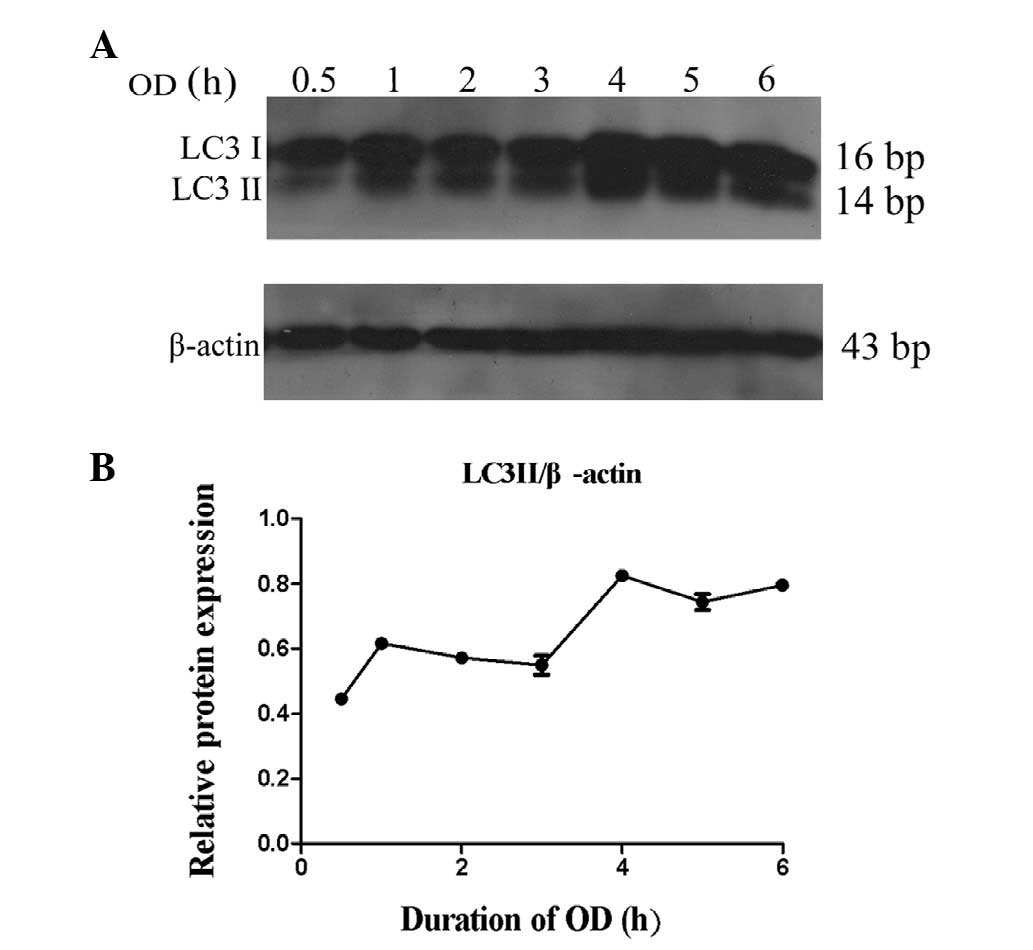
Induction of autophagy in OD neurons
In order to determine the timing of autophagy following OD treatment, the conversion of LC3 to 14 bp LC3II was analyzed. Total proteins were isolated from cultured neurons at 0.5, 1, 2, 3, 4, 5 and 6 h following OD treatment (n=3 per group). It was demonstrated that LC3 conversion began to increase at 0.5 h post-treatment, and significantly increased at 4–6 h (Fig. 1).
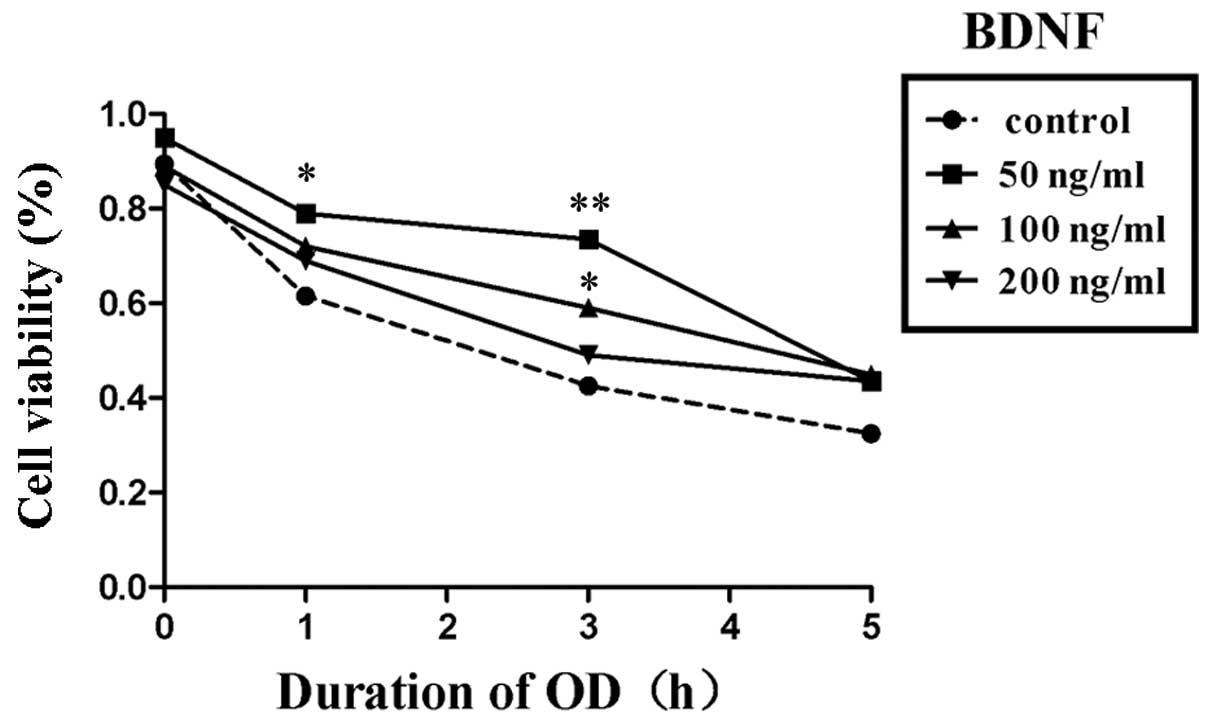
BDNF protects OD neurons by upregulating autophagy
To evaluate whether BDNF protected neurons from OD, the viability of cultured cortical neurons was detected by the CCK-8 assay. It was hypothesized that BDNF may exhibit autophagy activation in OD neurons similar to that of the autophagy promoter rapamycin.
Within the concentration range of 50 to 200 ng/ml, BDNF promoted cell viability compared with the negative control group. However, treatment with 50 ng/ml BDNF resulted in the greatest neuronal protection following OD for 5 h (Fig. 2).
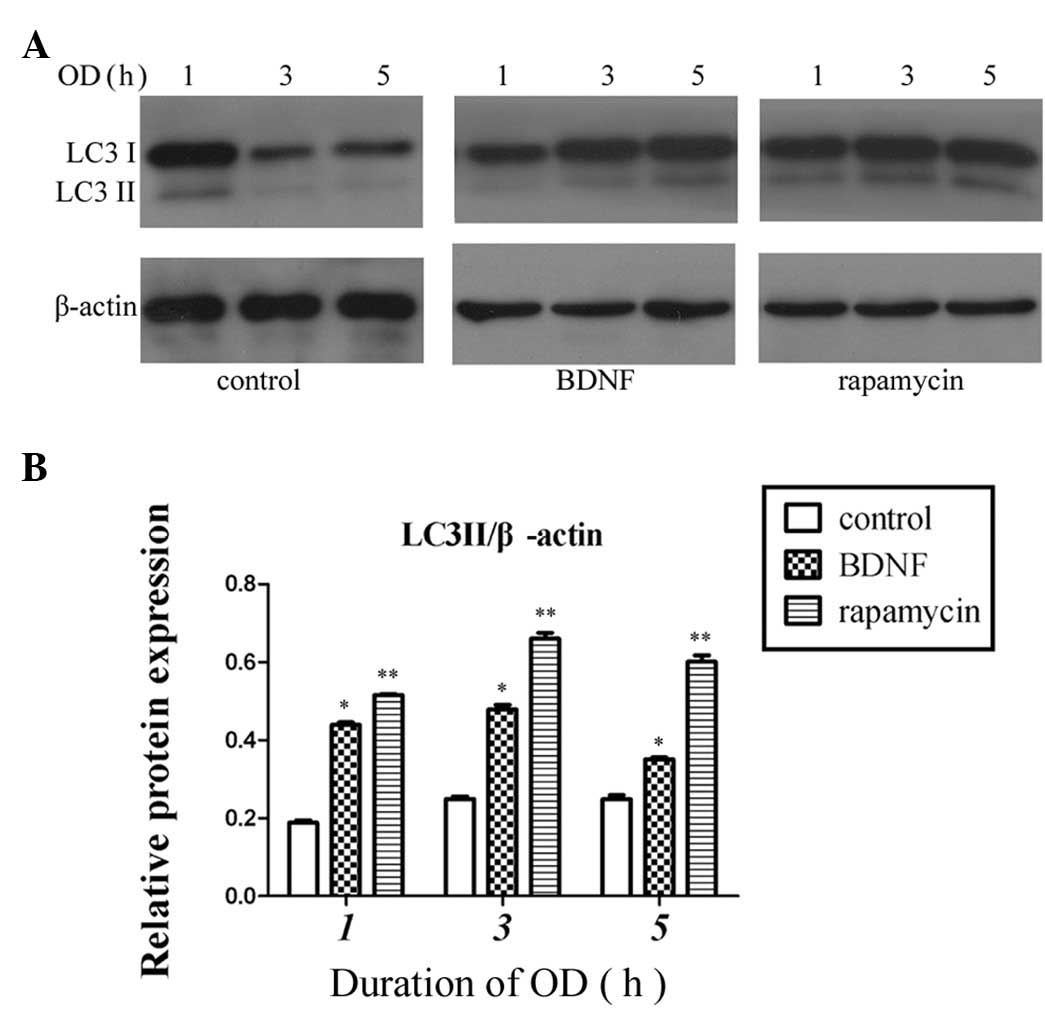
Therefore, combined with the CCK-8 assay results, 50 ng/ml BDNF was selected to be the interference concentration for subsequent experiments. BDNF (50 ng/ml) was added to the culture medium 24 h prior to OD and 300 nM rapamycin was added 30 min prior to OD, as in a previous study (8). Western blot analysis was used to detect the conversion of LC3. Compared with the control group, the BDNF and rapamycin groups upregulated the conversion of LC3; however, the expression of LC3 was greatest in the rapamycin group (P<0.01, Fig. 3).
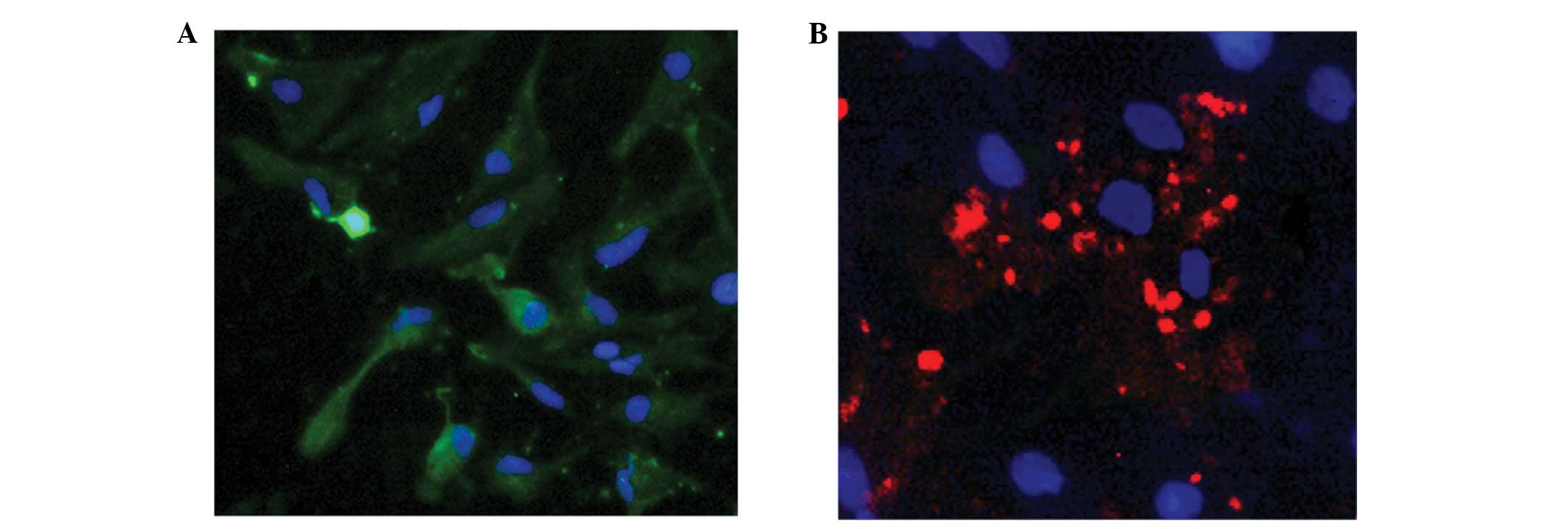
Immunofluorescence staining was used to analyze the formation of autophagosomes following BDNF treatment. MAP2 staining identified the cultured cells to be neurons (green; Fig. 4A), and LC3 aggregated in neurons with BDNF treatment, indicating the presence of autophagosomes (red; Fig. 4B).
BDNF induces the protection of OD neurons by enhancing autophagy via PI3K/Akt/mTOR/p70S6K inhibition
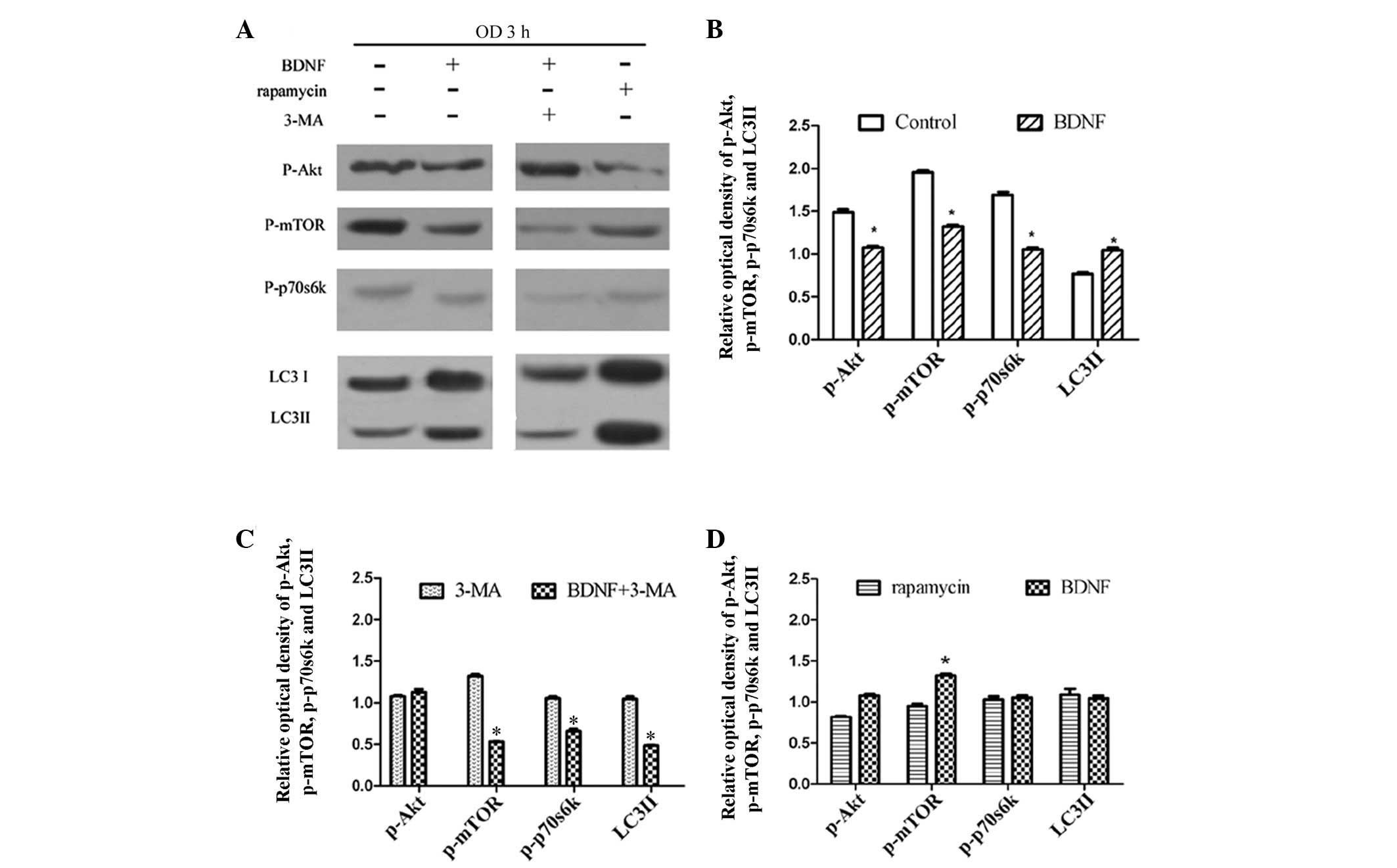
The PI3K/Akt/mTOR/p70S6K signaling pathway is regarded to be an important pathway involved in the regulation of autophagy. Moreover, it is also associated with neuroprotection and is often activated in ischemic brain damage. Our previous studies demonstrated that BDNF prevented cortical neurons from hypoxia-induced neurotoxicity through the activation of PI3K pathways, but not the p38 MAPK pathway (8). To investigate the signaling underlying the neuroprotection of BDNF against hypoxic insult, Sun et al(8) used LY294002 to inhibit PI3K. LY294002 partially blocked the neuroprotective effect of BDNF, suggesting that BDNF protected cultured cortical neurons from hypoxic injury via PI3K signaling pathways. To investigate whether BDNF upregulated autophagy in OD neurons through the PI3K/Akt/mTOR/p70S6K pathways, the level of p-Akt, p-mTOR, p-p70S6K and LC3 conversion were detected, respectively, by western blot analysis. Autophagy flux was also detected to confirm the activity of autophagy using the autophagy inhibitor, 3-methyladenine (3-MA).
At 3 h following OD treatment, BDNF pretreatment decreased the expression of p-Akt, p-mTOR and p-p70S6K, but increased the conversion of LC3 compared with the control group (P<0.05; Fig. 5A and B). In addition, 3-MA (10 mM) was used to inhibit autophagy induced by BDNF in OD neurons. As a result, it was demonstrated that following autophagy inhibitor 3-MA treatment, the level of p-mTOR, p-p70S6K and the conversion of LC3 upregulated by BDNF were decreased. Only the level of p-Akt was marginally increased (P>0.05; Fig. 5A). Furthermore, compared with the autophagy activator rapamycin, the levels of these proteins were similar with BDNF treatment (Fig. 5A and D).
Discussion
To the best of our knowledge, this study demonstrated for the first time that BDNF protected neurons from hypoxia injury in vitro via the activation of autophagy through the PI3K/Akt/mTOR/p70S6K signaling pathway.
Our previous studies demonstrated that the predominant neuropathology following ischemia/reperfusion (I/R) injury with the OGD model began during the acute insult and extended into the reperfusion phase (6–8). This study investigated ischemia but not reperfusion; therefore, OD was the only intervention that led to autophagy in the neurons. It was demonstrated that OD was capable of inducing autophagy in cultured neurons, suggesting that with a prolonged duration of OD, autophagy was upregulated to maintain homeostasis within the injured cells.
There are numerous controversies concerning the actual role of autophagy in cerebral ischemia depending on various pathological conditions neurons. A study demonstrated that pharmacological inhibition of autophagy resulted in attenuated focal cerebral ischemia-associated neural damage in rats (14). However, intracerebral ventricle injection of 3-MA following permanent occlusion of the middle cerebral artery (pMCAO) in rats significantly reduced infarct volume (15). In addition, the injury of ischemic brains in a rat model 3–7 days following ischemia was inhibited by the autophagy inhibitor, 3-MA (16). Moreover, Wang et al(17) determined that Nampt promoted neuronal survival through the induction of autophagy in MCAO rats. Furthermore, Balduini et al(18) observed that certain drugs with neuroprotective effects increased autophagy activity.
BDNF was discovered in 1982 and is a potent neuroprotective candidate that promotes the survival of neurons and may be a prospective treatment for neural injury induced by hypoxia. There are two types of BDNF, pro- and mature BDNF (19). Mature BDNF is released predominantly by neurons through constitutive secretion or in an activity-dependent manner, exerting its effect through the high-affinity receptor TrkB (6). Previous studies showed that BDNF was able to suppress apoptosis (20), inflammation, neurotoxicity (21) and promote neural regeneration (22). In addition, it also contributed to cognitive function and memory acquisition (23). However, few studies have investigated the involvement of autophagy in BDNF protection against hypoxia, thus this requires further investigation.
In the present study, the neuron viability was analyzed following treatment with BDNF and rapamycin (an autophagy activator). It was observed that, compared with the control group, cell viabilities in the 50–100 ng/ml BDNF and 300 nM rapamycin groups were elevated. BDNF upregulated autophagic activities similar to the action of rapamycin. The aggregation of LC3 was determined by an immunofluorescence assay. The LC3 particle was observed to be scattered in the neural plasma, surrounding the neural nuclei. These results suggested that moderate autophagy induced by BDNF may aid in protecting the cortical neurons against hypoxia in vitro.
Upon binding to TrkB, survival-promoting effects of BDNF are elicited by activating various intracellular signaling cascades, including the ERK and PI3K pathway. Akt, a 57 kDa serine/threonine kinase, is a predominant activator of the PI3K pathway. Once activated, signaling through Akt is propagated by a diverse array of substrates, including mTOR. mTOR is a serine/threonine protein kinase with a large molecular size (~300 kDa) that belongs to the phosphatidylinositol kinase-related kinase (PIK) family. It is widely accepted as a key gene/protein in the regulation of mammalian autophagy. Therefore, whether the association between BDNF and autophagy occurred through the PI3K/Akt/mTOR/p70S6K pathway was investigated.
Phosphorylated Akt, mTOR and p70S6K are the active forms of Akt, mTOR and p70S6K, respectively. In the present study, these proteins decreased markedly in the BDNF group, while the level of LC3II increased. Autophagy flux is another aspect of autophagy monitoring. 3-MA was used at a high concentration (10 mM) to inhibit the earliest stages of autophagosome formation (24). Therefore, 10 mM 3-MA was able to block the upregulation of LC3II induced by BDNF, and the levels of p-mTOR and p-70S6K declined. Data demonstrated that the Akt/mTOR/p70S6K pathway was interrupted by the autophagy inhibitor 3-MA.
However, p-mTOR and LC3 decreased simultaneously following 3-MA treatment. One possible reason is that 3-MA is a nonspecific inhibitor of class III PI3K. PI3Ks are a family of enzymes that are capable of phosphorylating phosphatidylinositol (PtdIns) at the 3′-hydroxyl group on the inositol ring. According to their specificity for the substrates and products, PI3Ks are categorized into three classes, class I, II and III. Class III PI3K is constitutively active and able to generate PtdIns(3)P from PtdIns. Upon activation, class I PI3K catalyzes PtdIns(4,5)P2 (the substrate for which it has the highest affinity) to produce PtdIns(3,4,5)P3, which triggers the downstream signaling cascade. mTOR binds several proteins to form two distinct protein complexes, mTORC1 and mTORC2. Akt binds to PtdIns(3,4,5)P3 via its pleckstrin homology (PH) domain and is phosphorylated. This p-Akt is further phosphorylated at serine 473 by mTOR complex II, which results in its full activation (25). Activated Akt results in the activation of mTOR complex I and the signals are passed to its downstream effector S6 kinase/ribosomal protein S6 (26,27). Therefore, 3-MA inhibited class III PI3K leading to the decline of p-Akt, p-mTOR and p-70S6K. No significant downregulation of p-Akt was identified, which is partly due to a negative feedback mechanism by which the downstream mTOR effector S6 kinase-1 (S6K1) also regulates the pathway by catalyzing the inhibitory phosphorylation of insulin receptor substrate (IRS) proteins. This prevents IRS proteins activating PI3K, thereby inhibiting the activation of Akt.
Rapamycin is an autophagy promoter and its preconditioning attenuates transient focal cerebral I/R injury (2,28) and increases axonal growth capacity of injured peripheral nerves in neuropathology (29). The beneficial effects of rapamycin to nerves were similar to that observed following treatment with BDNF (30). According to the results of the present study, changes in the levels of p-Akt, p-mTOR, p-p70S6 and LC3II following the addition of rapamycin were correlated with the action of BDNF in inducing autophagy (31).
Autophagy is an essential mechanism for maintaining cellular homeostasis when responding to stress. Our results revealed that autophagy was an important element of the endogenous defense mechanisms activated by OD preconditioning in vitro. BDNF may be considered as a novel neuroprotective candidate for its upregulation of autophagy. Although it was demonstrated that BDNF protected neurons from hypoxic injury by activation of autophagy through the PI3K/Akt/mTOR/p70S6K signaling pathway, further studies are required to confirm the specific effects and mechanism of BDNF.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant no. 30973215), the Science and Technology Pillar Projects of Sichuan Province (grant no. 2012SZ0010) and the Program for Changjiang Scholars and Innovative Research Team in University (grant no. IRT0935).
Abbreviations:
|
BDNF |
brain-derived neurotrophic factor |
|
PI3K |
phosphatidylinositol 3-kinase |
|
MAPK |
mitogen-activated protein kinase |
|
PLCγ |
phosphoinositide phospholipase C-γ |
|
CNS |
central nervous system |
|
HIE |
hypoxic-ischemic encephalopathy |
|
OD |
oxygen deprivation |
|
C3 |
caspase-3 |
|
CC3 |
cleaved caspase-3 |
|
LC3II |
microtubule-associated protein light chain 3 II |
|
p-Akt |
phosphorylated-Akt |
|
p-mTOR |
phosphorylated-mammalian target of rapamycin |
|
p-p70S6K |
phosphorylated-p70S6K |
|
HI |
hypoxic ischemic |
|
IV |
day in vitro |
|
PBS |
phosphate-buffered saline |
References
|
Klionsky DJ, Abdalla FC, Abeliovich H, et al: Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar | |
|
Carloni S, Girelli S, Scopa C, Buonocore G, Longini M and Balduini W: Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 6:366–377. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Yan W, Zhang H, Bai X, Lu Y, Dong H and Xiong L: Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 1402:109–121. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Lessmann V and Brigadski T: Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res. 65:11–22. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Massa SM, Yang T, Xie Y, et al: Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 120:1774–1785. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Meng M, Zhiling W, Hui Z, Shengfu L, Dan Y and Jiping H: Cellular levels of TrkB and MAPK in the neuroprotective role of BDNF for embryonic rat cortical neurons against hypoxia in vitro. Int J Dev Neurosci. 23:515–521. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Zhou H, Mao M, Liu W, Li S and Wang H: Expression of BDNF Receptor TrkBmRNA in hypoxia-induced fetal cortical neurons. J West China Univ Med Sci. 33:573–576. 2002. | |
|
Sun X, Zhou H, Luo X, et al: Neuroprotection of brain-derived neurotrophic factor against hypoxic injury in vitro requires activation of extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. Int J Dev Neurosci. 26:363–370. 2008. View Article : Google Scholar | |
|
Wu YT, Tan HL, Huang Q, Ong CN and Shen HM: Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 5:824–834. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Saiki S, Sasazawa Y, Imamichi Y, et al: Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Chen M, Du Y, Qui M, et al: Ophiopogonin B-induced autophagy in non-small cell lung cancer cells via inhibition of the PI3K/Akt signaling pathway. Oncol Rep. 29:430–436. 2013.PubMed/NCBI | |
|
Guo H, Ma J, Tong Y, Qu Y, Mu DZ and Mao M: A comparative study on three models of co-culture of neurons and astrocytes. Zhongguo Dang Dai Er Ke Za Zhi. 12:984–987. 2010.(In Chinese). | |
|
Zeng W, Tong Y, Li H, Luo R and Mao M: P2X7 receptor modulation of the viability of radial glial clone L2.3 cells during hypoxic-ischemic brain injury. Mol Med Rep. 5:1357–1361. 2012.PubMed/NCBI | |
|
Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H and Takeuchi T: Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 285:667–674. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Wen YD, Sheng R, Zhang LS, et al: Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 4:762–769. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and Kong J: Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 18:250–260. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Wang P, Guan YF, Du H, Zhai QW, Su DF and Miao CY: Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 8:77–87. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Balduini W, Carloni S and Buonocore G: Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med. 25(Suppl 1): 30–34. 2012. View Article : Google Scholar | |
|
Yang J, Siao CJ, Nagappan G, et al: Neuronal release of proBDNF. Nat Neurosci. 12:113–115. 2009. View Article : Google Scholar | |
|
Yao RQ, Qi DS, Yu HL, Liu J, Yang LH and Wu XX: Quercetin attenuates cell apoptosis in focal cerebral ischemia rat brain via activation of BDNF-TrkB-PI3K/Akt signaling pathway. Neurochem Res. 37:2777–2786. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Casalbore P, Barone I, Felsani A, D’Agnano I, Michetti F, Maira G and Cenciarelli C: Neural stem cells modified to express BDNF antagonize trimethyltin-induced neurotoxicity through PI3K/Akt and MAP kinase pathways. J Cell Physiol. 224:710–721. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Liang W, Han Q, Jin W, et al: The promotion of neurological recovery in the rat spinal cord crushed injury model by collagen-binding BDNF. Biomaterials. 31:8634–8641. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Blurton-Jones M, Kitazawa M, Martinez-Coria H, et al: Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA. 106:13594–13599. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Wu YT, Tan HL, Shui G, et al: Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
LoPiccolo J, Blumenthal GM, Bernstein WB and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Huang J and Manning BD: A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 37:217–222. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Maiese K, Chong ZZ, Shang YC and Wang S: mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 19:51–60. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Yin L, Ye S, Chen Z and Zeng Y: Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int J Neurosci. 122:748–756. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Abe N, Borson SH, Gambello MJ, Wang F and Cavalli V: Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. 285:28034–28043. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Chen AI, Xiong LJ, Tong Y and Mao M: The neuroprotective roles of BDNF in hypoxic ischemic brain injury (Review). Biomed Rep. 1:167–176. 2013.PubMed/NCBI | |
|
Yang YP, Liang ZQ, Gu ZL and Qin ZH: Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin. 26:1421–1434. 2005. View Article : Google Scholar : PubMed/NCBI |